Distinguishing Therapy-Induced Senescence and Polyploid Giant Cancer Cells: Impact on Breast Cancer Chemoresistance
bBiotechnology Program/RENORBIO, Health Sciences Center, Federal University of Espírito Santo, Vitória, Espírito Santo, Brazil,
cBiochemistry Program, Health Sciences Center, Federal University of Espírito Santo, Vitória, Espírito Santo, Brazil.
Keywords
Abstract
Cellular senescence and polyploid giant cancer cells (PGCCs) constitute distinct, yet interconnected, stress adaptation cellular programs that critically shape breast cancer (BC) progression and therapeutic response. Senescence arises via replicative, oncogene-induced, or therapy-induced mechanisms, and it is characterized by stable cell-cycle arrest and secretion of a senescence-associated secretory phenotype (SASP). Senescence can transiently suppress tumor growth; however, persistent senescence may ultimately facilitate immune evasion, cell survival, and chemoresistance. In turn PGCCs development comprises cytokinesis failure, endoreplication, or mitotic slippage and are notable for their enlarged morphology, genomic plasticity, and the ability to generate therapy-resistant progeny through atypical divisions. Both cell states often co-occur following genotoxic stress, as senescent cells can become polyploid and PGCCs may display senescence-associated features, complicating their distinction. While resistance mediated by therapy-induced senescence (TIS) is mainly driven by SASP signaling and reversible arrest, PGCC-driven resistance is associated with genetic diversification, acquisition of stemness, and long-term persistence. Therapeutic strategies include senolytics and senomorphics, as well as emerging PGCC-targeted approaches. By integrating morphological, molecular, and ploidy-based approaches to distinguish these phenotypes, this review aims to clarify their overlapping and divergent roles in BC chemoresistance and to highlight opportunities for more effective therapeutic interventions.
Introduction
Breast cancer (BC) is a heterogeneous malignant disease comprising multiple molecular subtypes that differ markedly in tumor development, progression, and therapeutic response [1]. Data from the American Cancer Society (ACS) ranks BC as the most prevalent cancer among women, accounting for 32% of diagnosed cases and 14% of cancer-related deaths anticipated in the United States for 2025. Additionally, the estimated incidence of invasive cancer is 2.1% among women under 49 years, increasing to 7.3% in those aged 65 to 84 years [2], pointing BC as an aging-related disease. This can be attributed to cumulative DNA damage, prolonged hormonal exposure, and immunosenescence, all of which contribute to a microenvironment conducive to tumorigenesis [3–5].
Beyond age, BC heterogeneity is largely driven by intrinsic molecular subtypes, which shape disease behavior and therapeutic outcomes. Immunohistochemical profiling based on estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) expression allows classification into four major clinical subtypes: luminal A (ER+ and/or PR+, HER2−), luminal B (ER+ and/or PR+, HER2±), HER2-enriched (ER−/PR−, HER2+), and triple-negative BC (TNBC; ER−/PR−/HER2−) [1, 6]. At the transcriptomic level, gene expression profiling further identifies intrinsic molecular subtypes, including basal-like tumors, which show substantial but incomplete overlap with TNBC. Basal-like tumors are typically characterized by high expression of basal cytokeratins 5, 6, and 14, EGFR, elevated proliferative index, genomic instability, and aggressive clinical behavior [6]. These subtypes exhibit distinct epidemiological patterns, biological aggressiveness, and responses to systemic therapy. Luminal A tumors, more prevalent among women over 50 years of age, are characterized by low proliferative index (Ki-67 <14%), favorable prognosis, and higher sensitivity to endocrine therapy [7]. In contrast, TNBC occurs more frequently in younger patients and is associated with high aggressiveness, early relapse, limited targeted treatment options, and variable responses to chemotherapy [8]. These age-related distributions and the subtype-specific differences underscore the clinical relevance of molecular classification in predicting therapy response and resistance.
Standard BC treatment includes surgery, chemotherapy, radiotherapy, endocrine therapy, and targeted agents, choices being guided by tumor subtype and disease stage [9]. Despite therapeutic advances, tumor heterogeneity, adverse effects, and multidrug resistance (MDR) continue to compromise treatment effectiveness. Chemoresistance, defined as the ability of cancer cells to evade cytotoxic agents, results from multiple interconnected mechanisms and represents a major challenge for successful management of BC metastatic disease [10]. Components of the tumor microenvironment (TME), especially factors secreted by cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs), facilitate MDR, while cancer stem cells (CSCs) contribute to drug resistance through their high tumorigenic potential and increased expression of ATP-binding cassette transporters [11].
Among cellular mechanisms underlying therapy resistance, therapy-induced senescence (TIS) has emerged as a critical adaptive response in BC. Senescence is defined as a stable cell-cycle arrest that protects against genomic instability and can be triggered by replicative exhaustion, DNA damage, or oxidative stress [12]. In cancer, therapeutic interventions such as chemotherapy, radiotherapy, and targeted therapy can induce this state, wherein cells cease proliferation but remain metabolically active and acquire a senescence-associated secretory phenotype (SASP), characterized by the release of cytokines, proteases, protease inhibitors, and growth-promoting factors [13]. Although TIS can initially exert antitumor effects by limiting the expansion of damaged or pre-malignant cells, SASP components also have persistent inflammation, angiogenesis, immune escape, and formation of a pro-tumorigenic microenvironment [14, 15]. Importantly, senescent cells may eventually evade growth arrest and resume proliferation. Large-scale screening of Food and Drug Administration (FDA)-approved anticancer agents has demonstrated that TIS confers resistance to nearly half of the tested compounds in multiple BC cell lines [16]. Collectively, these observations establish TIS as a reversible drug-resistant state that significantly promotes BC relapse and metastasis.
On the other hand, polyploid giant cancer cells (PGCCs) are distinguished from senescent cells by the presence of multiple nuclei or markedly enlarged nucleus [17]. Like TIS, PGCCs may arise from response to therapy-induced stress that promotes cell-cycle arrest or genomic alterations [18]. Their increased nuclear content and genomic redundancy enhance survival under cytotoxic pressure, strongly linking PGCCs to chemoresistance [19]. PGCCs and senescent cells share many characteristics: both can arrest cell-cycle, evade antineoplastic mechanisms, reorganize metabolism to ensure viability, and exhibit overlapping biomarkers, including senescence-associated β-galactosidase (SA-β-gal) activity [19]. This phenotypic overlap between senescent cells and PGCCs lacks selective molecular markers; therefore, compromising the distinction between TIS and PGCCs, challenging the choice of accurate BC treatment and the development of target therapeutic strategies. Given their overlapping features and distinct biological functions, deeper insights into the roles of TIS and PGCCs in chemoresistance, cellular aging, and BC progression are imperative. Although both phenomena originate from therapy-induced stress and share survival and evasion mechanisms, their outcomes, functions, and clinical implications considerably diverge. In this context, the present work aims to enlighten the differences between TIS and PGCCs, emphasizing how each state uniquely contributes to therapeutic resistance and tumor evolution, thereby assuring diagnostic interpretation and promoting the development of advanced BC targeted therapies.
Senescence versus polyploidization: distinct paths of cellular adaptation
Senescence, first described by Hayflick in 1961, is a defined cellular program characterized by profound morphological, biochemical, and metabolic remodeling that culminates in stable or long-lasting proliferative arrest [13, 20–22]. Despite loss of replicative capacity, senescent cells remain viable, metabolically active, and frequently resistant to apoptosis [23, 24]. Senescence is induced by diverse stressors, including genomic or telomeric damage, oxidative stress, epigenetic imbalance, oncogene activation, and tumor suppressor dysfunction, which converge to halt proliferation and prevent the propagation of genetically compromised cells [25, 26]. Among senescence subtypes, replicative senescence (RS) is a classical aging-associated barrier driven primarily by telomere attrition, mitochondrial dysfunction, and cumulative genomic instability [27–29]. RS functions as a potent tumor-suppressive mechanism, partly through activation of the senescence-associated secretory phenotype (SASP), which promotes immune-mediated clearance of damaged cells [27–29].
In the context of cancer biology, senescence can also be triggered by anticancer therapies [30], radiotherapy [31], endocrine therapy [21], or pathogen-associated stress [32]. These stimuli induce TIS or oncogene-induced senescence (OIS), which operate through distinct signaling pathways yet converge on a similar growth-arrested phenotype. The interplay among these mechanisms highlights senescence as both an intrinsic aging process and a treatment-modulated cellular outcome of relevance to BC [33].
Key drivers of RS
RS emerges when cumulative cellular stress exceeds the threshold required to maintain faithful replication, thereby enforcing irreversible growth arrest [34–37]. Mitochondrial dysfunction plays a central role in this process, arising from calcium overload, membrane depolarization, dysregulated permeability transition, and disruption of NAD⁺/NADH homeostasis [38, 39]. These alterations markedly increase reactive oxygen species (ROS) production, leading to oxidative damage of proteins, lipids, and nucleic acids and promoting genomic instability [34, 39, 40].
In parallel, telomere shortening constitutes a key aging-related mechanism leading to RS [33]. Progressive telomere erosion, inherent to lagging-strand DNA synthesis, compromises chromosomal end protection and elicits persistent DNA damage responses that drive senescence or apoptosis [41–44]. Genomic instability further reinforces RS, as age-related decline in DNA repair fidelity, checkpoint control, and telomere maintenance accelerates mutation accumulation and transcriptional dysregulation [45–49]. To counteract accumulating lesions, cells develop multiple DNA repair systems, including direct reversal, mismatch repair (MMR), nucleotide excision repair (NER), base excision repair (BER), homologous recombination (HR), and non-homologous end joining (NHEJ) [50]. MMR corrects base mismatches and insertion–deletion loops [51]: i) NER removes bulky adducts and transcription-blocking lesions [52]; ii) BER repairs oxidized or alkylated bases in nuclear and mitochondrial genomes [50, 53]; iii) and HR and NHEJ resolve single- and double-strand breaks [45]. When the extent of damage exceeds repair capacity, persistent lesions activate canonical senescence pathways: p53/p21/SDI1/CIP1 and p16INK4A/Rb, consolidating proliferative arrest [36, 54, 55].
The senescent phenotype evolves dynamically, progressing from early cell-cycle arrest to a fully established state marked by chromatin remodeling, lysosomal expansion, and robust SASP secretion [55–59]. While senescence initially supports tissue homeostasis and tumor suppression, chronic retention of senescent cells contributes to inflammation, tissue dysfunction, and age-related pathologies [60]. In breast tissue, intact RS machinery is therefore critical for restricting early tumorigenesis and shaping the cellular landscape upon which oncogenic events may later act [35, 46].
Key drivers of OIS
Oncogene-induced senescence (OIS) represents a mechanistically distinct yet convergent tumor-suppressive barrier triggered by aberrant oncogenic signaling rather than cumulative aging-related damage [13]. Unlike RS, OIS is acutely induced following excessive mitogenic stimulation that overwhelms cellular replication and repair capacity.
One of the best-characterized initiators of OIS is the oncogenic H-RASG12V variant, which normally regulates proliferation, differentiation, and survival. Mutations at codon 12 (G12V or G12D) lock RAS in a constitutively active state, driving excessive mitogenic signaling [61, 62]. This hyperproliferative status induces replicative stress signals and DNA damage, including fragile telomeres that are highly susceptible to breakage and persistent activation of the DNA damage response (DDR) [61, 63]. Sustained DDR signaling, in turn, engages canonical senescence pathways (p53/p21 and p16INK4A/Rb), enforcing cell-cycle arrest and stabilizing the senescent phenotype [54, 55].
Beyond RAS, dysregulation of PI3K/AKT and MAPK signaling, frequently observed in BC, can similarly trigger OIS when signaling intensity exceeds tolerable thresholds [62]. Effective execution of OIS critically depends on intact DDR and tumor suppressor pathways; failure of these checkpoints enables senescence bypass, genomic instability, and malignant progression [23]. Thus, while RS reflects a gradual aging-associated process and OIS an acute oncogenic stress response, both converge on shared molecular effectors to preserve genomic integrity and restrain tumor initiation.
Key drivers of TIS
TIS constitutes a clinically relevant, accelerated senescence program elicited by antineoplastic interventions, including chemotherapy, radiotherapy, and endocrine therapy [21, 30, 31]. These treatments induce substantial genotoxic and oxidative stress, increasing senescence-associated biomarkers, remodeling cellular morphology, and activating DDR signaling [30].
Mechanistically, most TIS-inducing agents converge on DNA damage, activating ATM/ATR kinases and downstream p53 signaling, thereby inhibiting cyclin–CDK complexes and enforcing growth arrest [64–66]. Parallel inhibition of CDKs by Chk1/Chk2 further consolidates this arrest, and senescence can arise even when individual DDR components are partially compromised [67]. TIS may also proceed through p16- and Rb-dependent mechanisms that block S-phase entry [68, 69].
In cancer, however, mutations affecting DDR components, ATM, or p53 may corrupt these checkpoints, enabling cells to evade or exit senescence [70]. Consequently, TIS may function not only as a growth-limiting response but also as a transient adaptive state that promotes survival under therapeutic stress and facilitates polyploidization, thereby contributing to the emergence of PGCCs and chemoresistance [16].
Key drivers of PGCCs
Polyploid cells, defined by the presence of more than two complete chromosome sets, occur physiologically in multiple tissues and contribute to development, regeneration, and differentiation [71–74]. In cancer, however, polyploidization represents a pathological adaptation frequently induced by genotoxic stress and anticancer therapies [75–77]. Cancer-associated polyploidy arises predominantly through endoreplication, cytokinesis failure, or cell fusion [73].
Endoreplication generates polyploid cells through repeated rounds of DNA synthesis without mitosis, a process favored by p53 and Rb dysfunction that disrupts cell-cycle control [73, 78, 79]. Cytokinesis failure and endomitosis similarly yield enlarged mono- or multinucleated cells with high DNA content [73, 79]. Cell fusion constitutes an additional route to polyploidization, involving coordinated stages of membrane remodeling, adhesion, and cytoplasmic merging, often resulting in highly unstable genomes [80–84].
PGCCs arise through these mechanisms and display senescence-like features, including growth arrest, metabolic remodeling, and resistance to apoptosis [79]. Importantly, PGCCs are typically transient, persisting for limited periods in vitro and in vivo, during which they enter a dormant yet viable state [85, 86]. Through reductive depolyploidization, PGCCs can generate genetically diverse diploid progeny with enhanced invasiveness, metastatic potential, and therapeutic resistance [79, 87]. Consistently, increased PGCC burden correlates with poor prognosis, advanced disease stage, and treatment failure across multiple tumor types, including BC [85, 88].
TIS and PGCCs: morphological and biomarker features
Distinguishing senescent cells from PGCCs remains challenging due to substantial phenotypic overlap. Senescent cells typically exhibit increased cell volume, flattened morphology, cytoplasmic vacuolization, and occasional multinucleation, reflecting sustained growth signaling in the absence of proliferation [60, 89]. PGCCs, by contrast, display proportional enlargement of both nucleus and cytoplasm, with markedly increased and irregular DNA-rich nuclei [79, 88, 90, 91].
From a molecular point of view, no single exclusive biomarker defines senescence; instead, a set of indicators is required. Cell-cycle arrest supports the senescence state when observed alongside suppressed expression of proliferation-associated genes in a pro-mitogenic context [92]. Increased lysosomal content, detectable as lipofuscin accumulation, and elevated SA-β-gal activity are still the most widely used markers in vitro and in vivo [60]. Expression of p16INK4a, p21Cip1/Waf1, and DDR components further supports senescence identification, although some of these markers may also be detectable at lower levels in PGCCs [93]. Several of these markers may also be detected in PGCCs, albeit in a context-dependent manner. PGCCs further express stemness-associated markers, including CD44 and CD133, and exhibit enhanced autophagic activity, reflecting shared stress-adaptive programs [80, 94].
Given the absence of exclusive molecular identifiers, quantitative assessment of ploidy and DNA content remains the most reliable discriminative criterion for PGCCs [91]. Recognizing the overlap and limitations of morphological and molecular markers is therefore essential for accurately interpreting therapy-induced cellular responses and understanding how genotoxic stress promotes the emergence of PGCCs in BC.
TIS and PGCCs in BC context
PGCCs have increasingly been recognized as critical contributors to BC progression, particularly in advanced disease, recurrence, and resistance to conventional therapies [18]. Clinically, PGCCs are frequently associated with metastatic dissemination and exhibit senescence-like features, reinforcing the phenotypic overlap between polyploidization and TIS [18].
Both preclinical and clinical evidence indicate that PGCCs emerge across multiple BC molecular subtypes. Analysis of human breast tumor biopsies encompassing luminal and basal-like tumors revealed the presence of PGCCs in both subtypes, with a higher abundance in basal-like tumors [95]. Notably, these samples displayed elevated levels of pro-inflammatory cytokines, suggesting an interaction between polyploidization and inflammatory signaling within the TME [95].
Despite their clinical relevance, there are currently no PGCC-targeted therapies in clinical trials [96]. Nonetheless, preclinical studies have begun to characterize therapy-induced PGCC formation (with CpCl2, a chemical hypoxia inducer, for example) and to identify compounds that selectively targets these cells, such as PRL3-zumab [87]. Taxanes such as docetaxel are commonly used to induce PGCCs in experimental models, enabling high-throughput drug screening approaches [96]. While most agents fail to eradicate PGCC populations, reflecting their intrinsic drug tolerance, selected compounds, including digoxin, disulfiram, azacitidine, decitabine, and zoledronic acid, have demonstrated efficacy against PGCCs in BC models [17, 87].
Several anticancer therapies have been shown to promote PGCC formation as an adaptive response to cytotoxic stress. Chemotherapeutic agents such as PARP inhibitors (olaparib and niraparib) and Aurora A kinase inhibitors (e.g., alisertib) induce polyploidization in BC in vitro models, accompanied by increased cell size, abnormal morphology, and expression of senescence-associated markers, including SA-β-gal, γ-H2AX, and p21 [62, 63, 97]. Importantly, subsets of PGCCs retain clonogenic potential and generate progeny cells, supporting the concept that polyploidization functions as a survival strategy enabling tumor repopulation following drug-induced stress [17, 18].
Antibody–drug conjugates (ADCs) have similarly been implicated in PGCC induction in BC. Agents such as trastuzumab emtansine, trastuzumab deruxtecan, XMT-1522, and disitamab vedotin generate PGCCs that subsequently give rise to smaller, low-DNA-content progeny consistent with a drug-tolerant persister phenotype [98]. Transcriptomic analyses of these cells reveal downregulation of proliferation-associated genes (e.g., MKI67 and CDK4) alongside with upregulation of cell-cycle arrest (GADD45A, p21), epithelial–mesenchymal transition (EMT), and autophagy-related pathways, coupled with reduced lamin B1 expression, a hallmark of senescence-like states [98].
Beyond therapeutic agents, environmental stressors may also promote PGCC formation. Exposure to the fungicide fludioxonil has been shown to induce polyploidization in the MDA-MB-231 BC cell line, characterized by increased cell size, multinucleation, impaired cell division, and elevated p53 expression, further emphasizing the sensitivity of cancer cells to stress-induced polyploid adaptation [99].
Collectively, current evidence supports PGCC formation as a common adaptive response to therapeutic and environmental stress in BC, strongly associated with chemoresistance, recurrence, and tumor progression [87]. Although clinical validation remains limited, ongoing preclinical research underscores the importance of elucidating PGCC biology to inform the development of targeted strategies aimed at improving treatment durability, reducing relapse, and ultimately enhancing patient survival.
TIS in BC chemoresistance and aging
Senescent cells are marked by extensive changes in chromatin organization, notably the formation of senescence-associated heterochromatin foci (SAHF), loss of lamin B1, and upregulation of CDK inhibitors p16INK4a and p21Cip1/Waf1 [100]. Additional hallmark features include SA-β-gal activity, persistent DNA damage foci, mitochondrial dysfunction, resistance to apoptosis, and stable cell-cycle arrest [15]. SASP is a defining characteristic of senescence, which has an impact on the development of age-related diseases and cancer [101]. In senescent cancer cells, SASP secretion comprises a mixture of cytokines, chemokines, growth factors, proteases, and extracellular matrix-modifying enzymes capable of reshaping the TME [102].
Although senescence initially acts as a tumor-suppressive mechanism by limiting the proliferation of damaged cells [15], mounting evidence highlights its paradoxical role in tumor progression and therapeutic resistance [103]. This duality is particularly evident in TIS, a cellular state elicited by chemotherapeutic agents, radiotherapy, and targeted therapies that induce DNA damage and oxidative stress, activating p53/p21 and p16/Rb signaling pathways [104]. TIS contributes to chemoresistance through multiple interconnected mechanisms, including sustained pro-survival signaling, evasion of apoptosis, and paracrine support mediated by SASP components such as IL-6, IL-8, and TGF-β [13, 16, 101, 105, 106]. These factors promote EMT-like traits, immune suppression, metastatic potential, and survival of neighboring cancer cells. Importantly, persistent senescent tumor cells can serve as a reservoir for tumor repopulation following therapy, contributing to residual disease and relapse, a phenomenon well documented in BC [13, 15, 100, 107].
Experimental studies in BC cell lines, including MCF-7, T47D, MDA-MB-231, and Hs578T, demonstrate that cells surviving chemotherapy frequently acquire senescence-associated features, such as p21 upregulation, γ-H2AX foci, BCL2L1 induction, and SA-β-gal positivity [16, 108]. Clinically, senescence-like phenotypes have been observed in tumors exhibiting incomplete pathological response to neoadjuvant chemotherapy [109]. In TNBC, chemotherapy-induced senescence is closely associated with stemness acquisition, SASP-mediated survival, and the emergence of drug-resistant clones [110]. Notably, senescence in cancer is not invariably terminal; a subset of senescent cells can escape growth arrest, re-enter the cell cycle, and generate progeny with enhanced aggressiveness and therapy resistance. This senescence escape involves transcriptional reprogramming that increases cellular plasticity and stem cell-like properties [16].
Beyond intrinsic survival advantages, senescent BC cells actively evade immune surveillance. Increased expression and altered glycosylation of PD-L1 have been observed during senescence, with ribophorin-1 implicated in PD-L1 processing [111]. In addition, elevated expression of DPP4/CD26 in senescent MCF-7 and MDA-MB-231 cells promotes immune escape and tumor cell survival. Pharmacological inhibition of DPP4 using sitagliptin, combined with the senolytic agent azithromycin, has shown synergistic efficacy in reducing senescent cell burden in vitro [112]. Notably, DPP4 is also recognized as a marker of aging, further linking senescence-associated immune modulation to age-related processes [113].
Critically, TIS shares extensive molecular and functional overlap with age-related senescence, including persistent DDR signaling, mitochondrial and metabolic dysfunction, chronic inflammation, and long-term SASP activity [57, 114]. In aged tissues, accumulation of senescent stromal, epithelial, and immune cells compromises immune surveillance and sustains a pro-tumorigenic microenvironment [115, 116]. In BC, these aging-associated alterations amplify the deleterious effects of TIS, increasing paracrine survival signaling and elevating the risk of treatment failure, particularly in elderly patients [117, 118].
Collectively, these findings underscore the central role of TIS in linking chemoresistance and aging in BC. Elucidating the mechanisms by which senescence promotes drug tolerance, including SASP signaling, immune evasion, and senescence escape, is essential for the development of effective therapeutic strategies. Incorporation of senolytic or senostatic agents into conventional treatment regimens holds promise for mitigating senescence-driven relapses and improving long-term patient outcomes.
PGCCs in BC chemoresistance and aging
PGCCs are increasingly recognized as an adaptive tumor cell state that can arise in response to severe cellular stress, particularly genotoxic insults induced by chemo- or radiotherapy [19, 79]. This subpopulation is characterized by whole-genome doubling or the accumulation of multiple chromosomal sets, resulting in markedly enlarged mono- or multinucleated cells with enhanced stress tolerance. Importantly, PGCCs have been shown to generate smaller, therapy-tolerant progeny through atypical division processes, suggesting a potential role in tumor persistence following treatment [17, 18]. Accumulating evidence indicate that increased PGCC abundance is associated with adverse clinical outcomes in breast, ovarian, and colorectal cancers, including disease progression, chemoresistance, metastasis, and recurrence [18, 19, 79].
PGCC formation can occur through multiple mechanisms, such as endoreplication, mitotic slippage, cytokinesis failure, cell fusion, and cell cannibalism [18, 79]. Microenvironmental stressors, including hypoxia, elevated reactive oxygen species, and persistent DNA damage, appear to further favor polyploidization [79, 80, 119]. Following induction, PGCCs often enter a transient dormant or slow-cycling state and may subsequently undergo neosis-like divisions or asymmetric budding, giving rise to progeny with increased genomic plasticity, stem-like features, and reduced sensitivity to anticancer therapies [80]. Consistent with this phenotype, PGCCs have been reported to express stemness-associated markers (e.g., CD44, OCT4, ALDH1A1, SOX2, NANOG, SSEA1), exhibit EMT-related traits, evade apoptosis partly via autophagy, and undergo metabolic reprogramming [120]. Single-cell transcriptomic analyses in BC further reveal that PGCCs display distinct cell-cycle regulation, ferroptosis susceptibility, and pronounced intrapopulation heterogeneity compared with non-polyploid tumor cells [17].
Notably, PGCCs share several features with senescent cells, including SA-β-gal activity, expression of γ-H2AX and p21, and secretion of pro-inflammatory cytokines such as IL-1β and IL-6 [6]. However, unlike terminally arrested senescent cells, PGCCs retain the capacity to exit dormancy and re-enter proliferative cycles, generating mitotically active and therapy-resistant progeny. This behavior has been observed in breast and ovarian cancer models exposed to DNA damage response inhibitors, including olaparib [6, 73, 120]. In addition, cytokines released by PGCCs may contribute to TME remodeling, supporting immune evasion, metastatic dissemination, and sustained drug tolerance [73, 120]. Collectively, these findings suggest that PGCC plasticity may facilitate post-treatment tumor relapse by combining stemness traits, senescence-like features, and lineage regeneration capacity [73, 121].
PGCC biology also intersects with aging-associated processes, including chronic oxidative stress, genomic instability, metabolic alterations, and activation of regenerative programs linked to stemness [73, 122, 123]. While polyploidization may represent an evolutionarily conserved stress-adaptation mechanism, malignant cells appear to exploit this plasticity to survive therapeutic pressure and repopulate tumors [80, 120].
In summary, both TIS and PGCC formation emerge as stress-adaptive responses to anticancer interventions but differ substantially in their temporal dynamics, molecular features, and biological consequences (Fig. 1). TIS is typically induced shortly after therapy and results in a growth-arrested, yet viable phenotype driven by p53/p21 or p16/Rb signaling, persistent DNA damage, and SASP secretion [104]. This state is associated with transient drug tolerance, paracrine support of neighboring cells, immune modulation, and the potential for later escape from arrest [16]. In contrast, PGCCs tend to arise following more prolonged or intense stress and contribute to longer-term resistance through genome reorganization, neosis-like division, acquisition of stemness features, and generation of genetically diverse progeny [18]. Although both states may express senescence-associated markers, PGCCs are uniquely defined by polyploidy, cellular gigantism, and extensive genomic plasticity, favoring lineage regeneration rather than stable arrest [17]. Distinguishing these adaptive programs has important implications for biomarker development, therapeutic targeting, and patient stratification, particularly in aging BC populations where clearance of stress-adapted cells may be compromised.
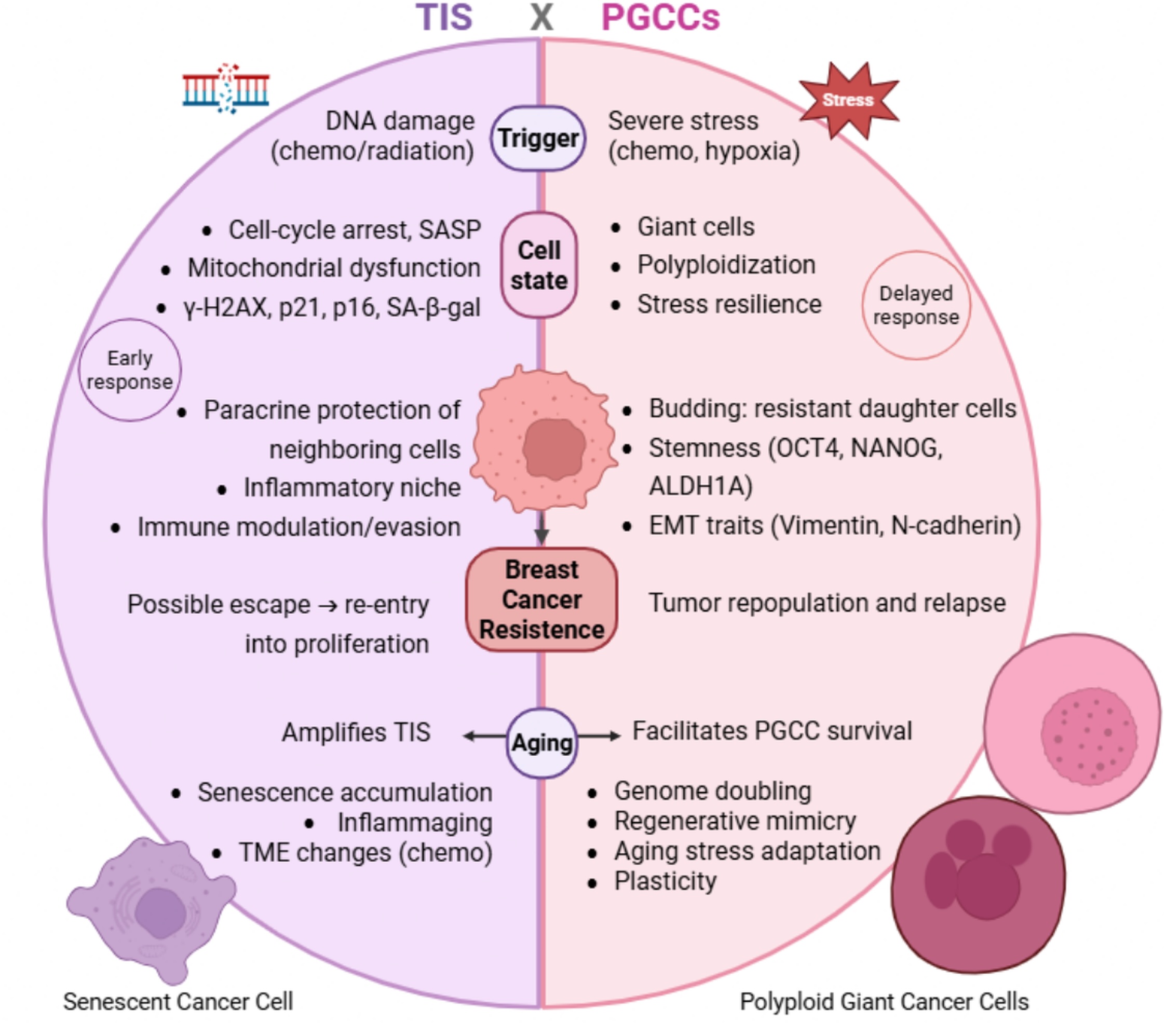
Fig. 1: Contrasting TIS- vs PGCCs- driven chemoresistance in BC. TIS typically occurs early post-therapy when cells suffer cycle arrest after DNA damage and adopt the senescent phenotype, with SASP secretion and survival advantages. Also, paracrine protection of nearby tumor cells, immune modulation, and escape from TIS and re-entry into proliferation after weeks. In contrast, PGCCs consist of a later adaptive response to extreme stress after therapy, involving polyploidization, stress resilience, stemness, EMT, and metastatic features, and the generation of progeny that work as a resource of resistant cells that contribute to tumor repopulation and relapse.
TIS and PGCCs therapeutic implications and clinical strategies in BC
Senescent cells may initially exert antitumor effects by enforcing stable cell-cycle arrest and limiting oncogenic signaling [13, 124]. For example, inhibition of NOTCH1 in murine TNBC models has been shown to induce senescence and enhance tumor responsiveness to immune checkpoint blockade, partly through SASP-mediated type I and II interferon signaling [125]. However, at later stages of disease, senescent cells can acquire protumorigenic functions that support tumor cell survival, therapy resistance, and disease progression, highlighting the context-dependent role of senescence in BC [13, 124].
Clinically, the accumulation of senescent cells has been associated with reduced sensitivity to CDK inhibitors, immune checkpoint inhibitors, and cytotoxic therapies. Importantly, the reversibility of TIS suggests potential therapeutic windows, as TIS-associated resistance is not always permanent [16]. In therapy-induced senescent BC cell lines (MCF-7, MDA-MB-231, Hs578T, and T47D), doxorubicin exposure led to senescence followed by partial loss of senescence markers, resumption of proliferation, and recovery of drug sensitivity [16]. In contrast, in HER2-enriched SKBR3 cells, escape from TIS was associated with increased chemoresistance [126]. These divergent outcomes indicate that the consequences of TIS reversal are influenced by tumor subtype, treatment context, and underlying genomic adaptations.
Accordingly, senotherapeutic strategies, including senolytics and senomorphics, have emerged as potential approaches to counteract senescence-associated resistance in BC [21, 127]. Senolytics selectively eliminate senescent cells by targeting anti-apoptotic pathways, such as BCL-2 family signaling [21], and include natural compounds (quercetin, fisetin), kinase inhibitors (dasatinib), and other targeted agents (e.g., RG7112, LBH589, OKI-179, ARV-825) [127, 128]. Preclinical studies report senolytic activity of compounds such as venetoclax, navitoclax, QD3, and fisetin in BC models [129–132], although clinical translation remains challenging for some agents due to toxicity or limited therapeutic windows [30]. In contrast, senomorphics aim to suppress SASP signaling and attenuate protumorigenic effects without eliminating senescent cells, commonly through inhibition of NF-κB or mTOR pathways [21].
Targeting PGCCs represents a complementary but still emerging therapeutic avenue in BC. Although these cells have been strongly implicated in therapy resistance and tumor relapse, selective strategies remain largely preclinical [17, 96]. Recent compound screens using 2D and 3D BC models identified proteasome inhibitors (bortezomib, carfilzomib, MG-132, ixazomib) and ferroptosis inducers (IKE, RSL3, FINO2, ML162, ML210) as active against PGCC-enriched populations across multiple BC subtypes, including TNBC and HER2-enriched tumors [17]. Additional candidates include CHK inhibitors (AZD7762, PF-477736) and the FOXM1 inhibitor thiostrepton, while drug repurposing screens have highlighted compounds such as the antimalarial pyranaridine [96].
The distinct biological properties of senescent cells and PGCCs necessitate differentiated therapeutic strategies, as illustrated in Fig. 2. While senescence may be induced by clinically tolerable drug doses, effective senolytic approaches often require higher or combination regimens that may increase systemic toxicity [30]. In contrast, advances in PGCC detection and high-throughput screening have accelerated the identification of candidate compounds, although in vivo validation and mechanistic elucidation remain ongoing. Overall, a refined understanding of the temporal dynamics and mechanistic differences between TIS- and PGCC-mediated resistance, particularly within aging TMEs, may inform rational combination strategies and support more personalized therapeutic interventions in BC.
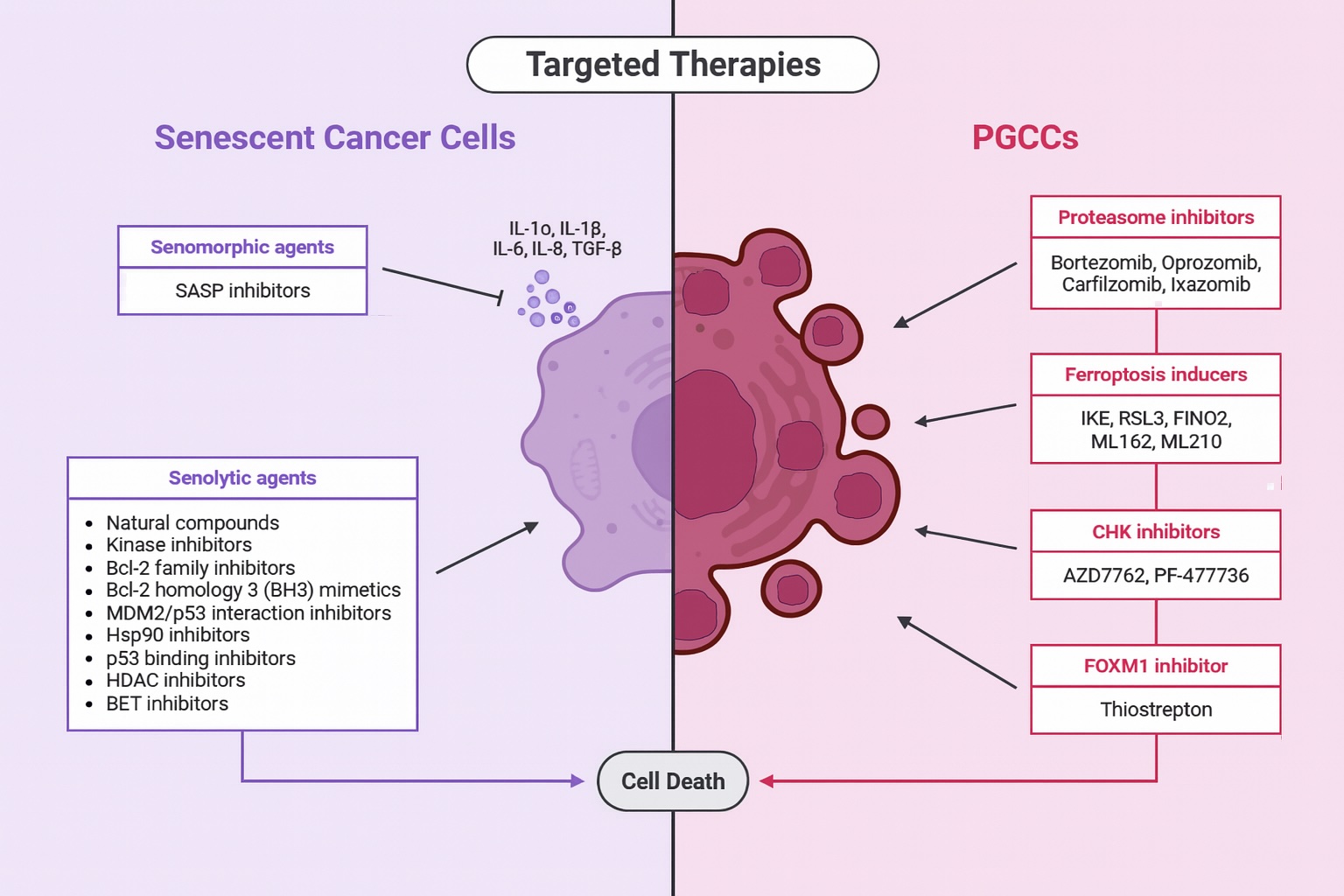
Fig. 2: Targeted therapies for senescent cells and PGCCs in BC. Targeted approaches exploit specific characteristics of each phenotype. In the context of senescent cells, therapeutic strategies include senomorphic and senolytic agents, which act through anti-apoptotic pathways, inhibition of SASP components, and induction of apoptosis. For PGCCs, targeted therapeutic agents include proteasome inhibitors, ferroptosis inducers, CHK and FOXM1 inhibitors, and other potential compounds. These agents represent a promising approach to overcoming chemoresistance and improving the clinical efficacy of treatment.
Experimental approaches to distinguish TIS from PGCCs in BC
TIS and PGCCs frequently emerge after genotoxic stress in BC and share overlapping phenotypic features, yet they represent biologically distinct stress-adapted states. Accurate discrimination between these phenotypes is essential for interpreting therapeutic responses, anticipating relapse risk, and understanding long-term tumor evolution [133]. Because no single biomarker reliably distinguishes TIS from PGCCs, current experimental strategies rely on multiparametric frameworks integrating functional, morphological, regulatory, and molecular readouts [134].
Contemporary approaches range from conventional immunocytochemistry and flow cytometry to advanced single-cell technologies, including high-resolution transcriptomic profiling [96]. Importantly, reliance on isolated markers, such as SA-β-gal or p16 alone, is discouraged, as these features may overlap in heterogeneous post-treatment populations [18, 71]. Instead, robust discrimination requires the coordinated assessment of DNA ploidy, proliferative status, DNA damage patterns, cell-cycle regulation, morphology, and transcriptional identity [96, 135].
Multiparametric integration
A multiparametric approach provides the most reliable distinction between TIS and PGCCs in BC by integrating complementary biological axes (summarized in Table 1) [19].

Table 1: Multiparametric framework for distinguishing TIS from PGCCs in BC. Parameters compiled across functional, regulatory, damage-related, morphological, stemness, and transcriptional axes to guide reliable phenotype identification
Quantification of DNA content by flow cytometry represents the most direct and objective discriminator. TIS typically remains within the 2N–4N range, even following substantial genotoxic stress, whereas PGCCs frequently exceed 8N because of mitotic failure or endoreduplication [18, 136]. This distinction has been consistently observed in BC models treated with paclitaxel, doxorubicin, or irradiation [122, 137]. Complementary evaluation of Ki-67 expression further refines classification: sustained Ki-67 suppression characterizes stable senescence, whereas PGCCs may transiently reactivate proliferation during neosis-like events, generating tumor-repopulating progeny [112, 135].
TIS is defined by robust and homogeneous induction of cell-cycle inhibitors, particularly p21, following treatment with CDK4/6 inhibitors or endocrine therapies in BC models [21, 138]. In contrast, PGCCs often display irregular, transient, or reduced p21 and p16 expression, reflecting an inability to maintain stable arrest. Loss or dysfunction of p53 further facilitates polyploidization and proliferative escape [136, 137]. When combined with ploidy analysis, immunocytochemical detection of these regulators substantially strengthens phenotype classification.
The spatial organization of DNA damage provides an additional discriminatory layer. In TIS, residual lesions typically appear as discrete, persistent γ-H2AX foci, reflecting stabilized DNA damage signaling [5, 21]. PGCCs, by contrast, often exhibit diffuse and intense γ-H2AX staining, consistent with ongoing chromosomal instability and defective mitosis during polyploidization [122, 137]. Thus, both the presence and distribution of γ-H2AX signals are informative for distinguishing stabilized senescence from progressive genomic destabilization.
Morphological assessment remains a powerful and accessible discriminator. PGCCs display hallmark features such as extreme cell enlargement, multinucleation, irregular chromatin organization, prominent nucleoli, and evidence of aborted cytokinesis, frequently linked to mitotic stress or Aurora kinase inhibition [97, 136, 137]. TIS cells, in contrast, exhibit moderate enlargement without overt mitotic catastrophe, reinforcing morphology as a critical classification parameter when interpreted alongside ploidy.
PGCCs and their progeny commonly exhibit elevated ALDH activity and a CD44high/CD24low phenotype, consistent with enhanced regenerative capacity and therapeutic resistance [122, 136]. TIS cells generally lack these features and show diminished self-renewal potential [17, 21]. This axis is particularly informative for identifying proliferative escape events driven by PGCC-derived lineages after therapy.
Single-cell RNA sequencing provides the highest-resolution discrimination between these states. TIS is characterized by SASP-enriched transcriptional programs, global suppression of cell-cycle genes, and strong p21-associated signaling. In contrast, PGCCs display gene expression profiles linked to cellular plasticity, metabolic rewiring, regenerative potential, and drug tolerance [17, 96, 112]. Integration of transcriptomic, morphological, and functional data confirms that PGCCs represent a minor yet biologically impactful subpopulation contributing to therapeutic resistance and tumor recurrence in BC.
Individually, each parameter provides only partial discrimination. When combined, however, they establish a high-precision analytical framework: ploidy defines genomic context; p21 and Ki-67 indicate proliferative stability; γ-H2AX patterns reflect DNA damage dynamics; morphology captures mitotic integrity; stemness markers reveal regenerative capacity; and transcriptomics resolves molecular identity [139–143]. Together, these axes offer the most reliable strategy for distinguishing TIS from PGCC-associated phenotypes, with direct implications for experimental interpretation and therapeutic decision-making in BC.
Conclusion
Accumulating evidence indicates that BC progression and therapeutic failure are tightly intertwined with aging-associated biological processes. Cellular senescence, chronic SASP signaling, and the emergence of PGCCs collectively shape a tumor ecosystem characterized by persistent inflammation, metabolic rewiring, and enhanced survival rates under stress. These aging-driven alterations not only promote adaptability and resistance to endocrine or chemotherapeutic agents but also generate a reservoir of highly plastic cells capable of driving tumor recurrence and metastasis. Recognizing how aging reprograms cancer cell fate is essential for redefining vulnerability nodes in BC. This review proposes that targeting aging1 related mechanisms, particularly those governing senescence and PGCC formation, may be pivotal for restoring therapeutic sensitivity and improving long-term outcomes in BC.
Acknowledgements
The authors would like to thank the institutional support provided by the Federal University of Espírito Santo (UFES). The authors also recognize the support of Brazilian funding agencies, including CAPES, CNPq, and FAPES.
Author Contributions
T.M.P. conceived the study, designed the review structure, conducted the literature search, performed data
analysis and interpretation, prepared primary drafts, translated and revised the manuscript, and carried
out all
critical revisions. B.S.M. contributed to conceptual organization, assisted in manuscript drafting, and
provided
methodological support by training the team in the use of reference management software. J.C.S., M.G.M.,
C.M.R.,
L.G.T., S.M.S.B., J.M.S.P, and L.B.B. contributed to writing specific subsections and assisting with
figure or
table preparation. L.B.A.R. supervised the project, provided critical intellectual input throughout the
development of the manuscript, ensured scientific accuracy, and approved the final version of the work.
Funding Sources
The authors declare that no specific external funding was received for this study.
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors declare no conflicts of interest. Artificial intelligence tools (ChatGPT, OpenAI; and Perplexity AI) were used exclusively to support English language editing, refinement and clarity, without influencing the scientific content, data interpretation, or conclusions. All conceptualization, analysis, interpretation of the literature, writing of scientific content, and final approval of the manuscript were performed by the authors.
References
| 1 | Xiong X, Zheng LW, Ding Y, Chen YF, Cai YW, Wang LP, Huang L, Liu CC, Shao ZM, Yu K Da. Breast
cancer: pathogenesis and treatments. Signal Transduction and Targeted Therapy 2025 10:1 [Internet].
Nature Publishing Group; 2025 [cited 2025 Nov 10]; 10: 1-33.
https://doi.org/10.1038/s41392-024-02108-4 |
| 2 | Siegel RL, Kratzer TB, Giaquinto AN, Sung H, Jemal A. Cancer statistics, 2025 Ca [Internet].
Wiley; 2025 [cited 2025 Nov 10]; 75: 10.
https://doi.org/10.3322/caac.21871 |
| 3 | Hamajima N, Hirose K, Tajima K, Rohan T, Friedenreich CM, Gapstur SM, Gaudet MM, Coates RJ, Liff
JM, Kakouri E, Marcou Y, Duffy SW, Morabia A, et al. Type and timing of menopausal hormone therapy
and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological
evidence. Lancet [Internet]. Elsevier B.V.; 2019 [cited 2025 Nov 10]; 394: 1159.
https://doi.org/10.1016/S0140-6736(19)31709-X |
| 4 | Fu Z, Xu H, Yue L, Zheng W, Pan L, Gao F, Liu X. Immunosenescence and cancer: Opportunities and
challenges. Medicine [Internet]. Lippincott Williams and Wilkins; 2023 [cited 2025 Nov 10]; 102:
e36045.
https://doi.org/10.1097/MD.0000000000036045 |
| 5 | Anglada T, Repullés J, Espinal A, LaBarge MA, Stampfer MR, Genescà A, Martín M. Delayed γH2AX foci
disappearance in mammary epithelial cells from aged women reveals an age-associated DNA repair
defect. Aging [Internet]. Impact Journals, LLC; 2019 [cited 2025 Nov 10]; 11: 1510-23.
https://doi.org/10.18632/aging.101849 |
| 6 | Zhang X. Molecular Classification of Breast Cancer: Relevance and Challenges. Arch Pathol Lab Med
[Internet]. Allen Press; 2023 [cited 2025 Nov 13]; 147: 46-51.
https://doi.org/10.5858/arpa.2022-0070-RA |
| 7 | Wongmaneerung P, Chitapanarux I, Traisathit P, Prasitwattanaseree S, Rottuntikarn W,
Somwangprasert A, Ditsatham C, Watcharachan K, Klunklin P, Onchan W. The association between Ki-67
expression and survival in breast cancer subtypes: a cross-sectional study of Ki-67 cut-point in
northern Thailand. BMC Cancer [Internet]. BioMed Central Ltd; 2025 [cited 2025 Nov 13]; 25: 346.
https://doi.org/10.1186/s12885-025-13724-w |
| 8 | Kumar N, Ehsan S, Banerjee S, Perez CF, Lhuilier I, Neuner J, Friebel-Klingner T, Fayanju OM, Nair
B, Niinuma SA, Nampoothiri S, McCarthy AM. The unique risk factor profile of triple-negative breast
cancer: a comprehensive meta-analysis. JNCI Journal of the National Cancer Institute [Internet].
Oxford University Press; 2024 [cited 2025 Nov 13]; 116: 1210.
https://doi.org/10.1093/jnci/djae056 |
| 9 | Mumtaz S, Ali S, Mumtaz S, Pervaiz A, Tahir HM, Farooq MA, Mughal TA. Advanced treatment
strategies in breast cancer: A comprehensive mechanistic review. Sci Prog [Internet]. SAGE
Publications Ltd; 2023 [cited 2025 Nov 13]; 106: 00368504231175331.
https://doi.org/10.1177/00368504231175331 |
| 10 | Singh S, Saini H, Sharma A, Gupta S, Huddar VG, Tripathi R. Breast cancer: miRNAs monitoring
chemoresistance and systemic therapy. Front Oncol [Internet]. Frontiers Media SA; 2023 [cited 2025
Nov 13]; 13: 1155254.
https://doi.org/10.3389/fonc.2023.1155254 |
| 11 | Ahmadpour ST, Orre C, Bertevello PS, Mirebeau-Prunier D, Dumas JF, Desquiret-Dumas V. Breast
Cancer Chemoresistance: Insights into the Regulatory Role of lncRNA. Int J Mol Sci [Internet].
Multidisciplinary Digital Publishing Institute (MDPI); 2023 [cited 2025 Nov 13]; 24: 15897.
https://doi.org/10.3390/ijms242115897 |
| 12 | Saleh T, Carpenter VJ, Bloukh S, Gewirtz DA. Targeting Tumor Cell Senescence and Polyploidy as
Potential Therapeutic Strategies. Semin Cancer Biol [Internet]. Academic Press; 2020 [cited 2025 Nov
15]; 81: 37.
https://doi.org/10.1016/j.semcancer.2020.12.010 |
| 13 | Chembukavu SN, Lindsay AJ. Therapy-induced senescence in breast cancer: an overview. Explor Target
Antitumor Ther [Internet]. Open Exploration Publishing Inc; 2024 [cited 2025 Nov 14]; 5: 902.
https://doi.org/10.37349/etat.2024.00254 |
| 14 | Schmitt CA, Wang B, Demaria M. Senescence and cancer - role and therapeutic opportunities. Nat Rev
Clin Oncol [Internet]. Springer Nature; 2022 [cited 2025 Nov 15]; 19: 619.
https://doi.org/10.1038/s41571-022-00668-4 |
| 15 | Luo J, Sun T, Liu Z, Liu Y, Liu J, Wang S, Shi X, Zhou H. Persistent accumulation of
therapy-induced senescent cells: an obstacle to long-term cancer treatment efficacy. International
Journal of Oral Science 2025 17:1 [Internet]. Nature Publishing Group; 2025 [cited 2025 Nov 15]; 17:
59-.
https://doi.org/10.1038/s41368-025-00380-w |
| 16 | Bajtai E, Kiss C, Bakos É, Langó T, Lovrics A, Schád É, Tisza V, Hegedűs K, Fürjes P, Szabó Z,
Tusnády GE, Szakács G, Tantos Á, et al. Therapy-induced senescence is a transient drug resistance
mechanism in breast cancer. Mol Cancer [Internet]. BioMed Central Ltd; 2025 [cited 2025 Nov 15]; 24:
128.
|
| 17 | Zhou M, Ma Y, Chiang CC, Rock EC, Butler SC, Anne R, Yatsenko S, Gong Y, Chen YC. Single-cell
morphological and transcriptome analysis unveil inhibitors of polyploid giant breast cancer cells in
vitro. Commun Biol [Internet]. Nature Research; 2023 [cited 2025 Nov 14]; 6: 1301.
https://doi.org/10.1038/s42003-023-05674-5 |
| 18 | Saini G, Joshi S, Garlapati C, Li H, Kong J, Krishnamurthy J, Reid MD, Aneja R. Polyploid giant
cancer cell characterization: New frontiers in predicting response to chemotherapy in breast cancer.
Semin Cancer Biol [Internet]. Academic Press; 2021 [cited 2025 Nov 15]; 81: 220.
https://doi.org/10.1016/j.semcancer.2021.03.017 |
| 19 | Ogawa Y, Fisher L, Matsumoto T. The Impact of Polyploid Giant Cancer Cells: The Root of Stress
Resilience. Cancer Sci [Internet]. John Wiley and Sons Inc; 2025 [cited 2025 Nov 15]; 116: 2949.
https://doi.org/10.1111/cas.70191 |
| 20 | Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res
[Internet]. Exp Cell Res; 1961 [cited 2025 Nov 26]; 25: 585-621.
https://doi.org/10.1016/0014-4827(61)90192-6 |
| 21 | McGrath MK, Abolhassani A, Guy L, Elshazly AM, Barrett JT, Mivechi NF, Gewirtz DA, Schoenlein P V.
Autophagy and senescence facilitate the development of antiestrogen resistance in ER positive breast
cancer. Front Endocrinol (Lausanne). Frontiers Media SA; 2024; 15.
https://doi.org/10.3389/fendo.2024.1298423 |
| 22 | Tiwari P, Shukla SK, Rastogi Verma S. Unraveling senescence in cancer: mechanistic complexities
and therapeutic opportunities. Molecular Biology Reports 2025 52:1 [Internet]. Springer; 2025 [cited
2025 Nov 11]; 52: 1-13.
https://doi.org/10.1007/s11033-025-10630-z |
| 23 | Roger L, Tomas F, Gire V. Mechanisms and Regulation of Cellular Senescence. Int J Mol Sci
[Internet]. MDPI; 2021 [cited 2025 Nov 11]; 22: 13173.
https://doi.org/10.3390/ijms222313173 |
| 24 | Gergues M, Bari R, Koppisetti S, Gosiewska A, Kang L, Hariri RJ. Senescence, NK cells, and cancer:
navigating the crossroads of aging and disease. Front Immunol [Internet]. Frontiers Media SA; 2025
[cited 2025 Nov 11]; 16: 1565278.
https://doi.org/10.3389/fimmu.2025.1565278 |
| 25 | Kepp O, Galluzzi L, Petroni G. Cellular senescence and aging at the crossroad between immunity and
cancer. Methods Cell Biol [Internet]. Academic Press; 2024 [cited 2025 Nov 11]; 181: xvii-xxiv.
https://doi.org/10.1016/S0091-679X(24)00009-8 |
| 26 | Wang L, Lankhorst L, Bernards R. Exploiting senescence for the treatment of cancer. Nature Reviews
Cancer 2022 22:6 [Internet]. Nature Publishing Group; 2022 [cited 2025 Nov 11]; 22: 340-55.
https://doi.org/10.1038/s41568-022-00450-9 |
| 27 | Talukdar S, Das SK, Emdad L, Fisher PB. Autophagy and senescence: Insights from normal and cancer
stem cells. Adv Cancer Res [Internet]. Academic Press; 2021 [cited 2025 Nov 11]; 150: 147-208.
https://doi.org/10.1016/bs.acr.2021.01.005 |
| 28 | González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D. A guide to assessing cellular senescence in
vitro and in vivo. FEBS Journal [Internet]. Blackwell Publishing Ltd; 2021 [cited 2025 Nov 11]; 288:
56-80.
https://doi.org/10.1111/febs.15570 |
| 29 | Shimizu K, Inuzuka H, Tokunaga F. The interplay between cell death and senescence in cancer. Semin
Cancer Biol [Internet]. Academic Press; 2025 [cited 2025 Nov 11]; 108: 1-16.
https://doi.org/10.1016/j.semcancer.2024.11.001 |
| 30 | de Paula B, Kieran R, Koh SSY, Crocamo S, Abdelhay E, Muñoz-Espín D. Targeting Senescence as a
Therapeutic Opportunity for Triple-Negative Breast Cancer. Mol Cancer Ther. American Association for
Cancer Research Inc.; 2023; 22: 583-98.
https://doi.org/10.1158/1535-7163.MCT-22-0643 |
| 31 | Tabasso AFS, Jones DJL, Jones GDD, Macip S. Radiotherapy-Induced Senescence and its Effects on
Responses to Treatment. Clin Oncol (R Coll Radiol) [Internet]. Clin Oncol (R Coll Radiol); 2019
[cited 2025 Nov 10]; 31: 283-9.
https://doi.org/10.1016/j.clon.2019.02.003 |
| 32 | McHugh D, Durán I, Gil J. Senescence as a therapeutic target in cancer and age-related diseases.
Nature Reviews Drug Discovery 2024 24:1 [Internet]. Nature Publishing Group; 2024 [cited 2025 Nov
11]; 24: 57-71.
https://doi.org/10.1038/s41573-024-01074-4 |
| 33 | Billimoria R, Bhatt P. Senescence in cancer: Advances in detection and treatment modalities.
Biochem Pharmacol [Internet]. Elsevier; 2023 [cited 2025 Nov 11]; 215: 115739.
https://doi.org/10.1016/j.bcp.2023.115739 |
| 34 | Martini H, Passos JF. Cellular senescence: all roads lead to mitochondria. FEBS J [Internet]. John
Wiley and Sons Inc; 2022 [cited 2025 Nov 12]; 290: 1186.
https://doi.org/10.1111/febs.16361 |
| 35 | Pizzul P, Rinaldi C, Bonetti D. The multistep path to replicative senescence onset: zooming on
triggering and inhibitory events at telomeric DNA. Front Cell Dev Biol [Internet]. Frontiers Media
SA; 2023 [cited 2025 Nov 12]; 11: 1250264.
https://doi.org/10.3389/fcell.2023.1250264 |
| 36 | Chesnokova V, Melmed S. GH and Senescence: A New Understanding of Adult GH Action. J Endocr Soc
[Internet]. J Endocr Soc; 2021 [cited 2025 Nov 12]; 6.
https://doi.org/10.1210/jendso/bvab177 |
| 37 | Revy P, Kannengiesser C, Bertuch AA. Genetics of human telomere biology disorders. Nature Reviews
Genetics 2022 24:2 [Internet]. Nature Publishing Group; 2022 [cited 2025 Nov 12]; 24: 86-108.
https://doi.org/10.1038/s41576-022-00527-z |
| 38 | Escrig-Larena JI, Delgado-Pulido S, Mittelbrunn M. Mitochondria during T cell aging. Semin Immunol
[Internet]. Academic Press; 2023 [cited 2025 Nov 12]; 69: 101808.
https://doi.org/10.1016/j.smim.2023.101808 |
| 39 | Miwa S, Kashyap S, Chini E, von Zglinicki T. Mitochondrial dysfunction in cell senescence and
aging. J Clin Invest [Internet]. American Society for Clinical Investigation; 2022 [cited 2025 Nov
12]; 132: e158447.
https://doi.org/10.1172/JCI158447 |
| 40 | Gao X, Yu X, Zhang C, Wang Y, Sun Y, Sun H, Zhang H, Shi Y, He X. Telomeres and Mitochondrial
Metabolism: Implications for Cellular Senescence and Age-related Diseases. Stem Cell Rev Rep
[Internet]. Springer; 2022 [cited 2025 Nov 12]; 18: 2315.
https://doi.org/10.1007/s12015-022-10370-8 |
| 41 | Yu L, Kang Y, Yu M, Ding Y, Chen Y, Wu Q, Cai Y, Liu H, Lv Z. Senescence-Inducing Therapy
Sequential NIR-II Mild Photothermal/Senolytic Therapy of Triple Negative Breast Cancer. Advanced
Science. John Wiley and Sons Inc; 2025.
https://doi.org/10.1002/advs.202507248 |
| 42 | Rossiello F, Jurk D, Passos JF, d'Adda di Fagagna F. Telomere dysfunction in ageing and
age-related diseases. Nat Cell Biol [Internet]. Nature Research; 2022 [cited 2025 Nov 13]; 24: 135.
https://doi.org/10.1038/s41556-022-00842-x |
| 43 | Lin J, Epel E. Stress and telomere shortening: Insights from cellular mechanisms. Ageing Res Rev
[Internet]. Elsevier Ireland Ltd; 2021 [cited 2025 Nov 16]; 73: 101507.
https://doi.org/10.1016/j.arr.2021.101507 |
| 44 | Chakravarti D, LaBella KA, DePinho RA. Telomeres: History, Health and Hallmarks of Aging. Cell
[Internet]. Cell Press; 2021 [cited 2025 Nov 17]; 184: 306.
https://doi.org/10.1016/j.cell.2020.12.028 |
| 45 | Wu H, Roks AJM. Genomic instability and vascular aging: A focus on nucleotide excision repair.
Trends Cardiovasc Med [Internet]. Trends Cardiovasc Med; 2014 [cited 2025 Nov 26]; 24: 61-8.
https://doi.org/10.1016/j.tcm.2013.06.005 |
| 46 | López-Otín C, Pietrocola F, Roiz-Valle D, Galluzzi L, Kroemer G. Meta-hallmarks of aging and
cancer. Cell Metab [Internet]. Cell Press; 2023 [cited 2025 Nov 26]; 35: 12-35.
https://doi.org/10.1016/j.cmet.2022.11.001 |
| 47 | Niedernhofer LJ, Gurkar AU, Wang Y, Vijg J, Hoeijmakers JHJ, Robbins PD. Nuclear Genomic
Instability and Aging. Annu Rev Biochem [Internet]. Annu Rev Biochem; 2018 [cited 2025 Nov 18]; 87:
295-322.
https://doi.org/10.1146/annurev-biochem-062917-012239 |
| 48 | Zhivotovsky B, Kroemer G. Apoptosis and genomic instability. Nat Rev Mol Cell Biol [Internet]. Nat
Rev Mol Cell Biol; 2004 [cited 2025 Nov 26]; 5: 752-62.
https://doi.org/10.1038/nrm1443 |
| 49 | Graziano S, Gonzalo S. Mechanisms of oncogene-induced genomic instability. Biophys Chem
[Internet]. Elsevier B.V.; 2017 [cited 2025 Nov 26]; 225: 49-57.
https://doi.org/10.1016/j.bpc.2016.11.008 |
| 50 | Popov AA, Petruseva IO, Lavrik OI. Activity of DNA Repair Systems in the Cells of Long-Lived
Rodents and Bats. Biochemistry (Moscow). 2024; 89: 1014-23.
https://doi.org/10.1134/S0006297924060038 |
| 51 | Mestrallet G, Brown M, Bozkus CC, Bhardwaj N. Immune escape and resistance to immunotherapy in
mismatch repair deficient tumors. Front Immunol [Internet]. Frontiers Media SA; 2023 [cited 2025 Nov
21]; 14: 1210164.
https://doi.org/10.3389/fimmu.2023.1210164 |
| 52 | Rechkunova NI, Krasikova YS, Lavrik OI. Nucleotide excision repair: DNA damage recognition and
preincision complex assembly. Biochemistry (Mosc) [Internet]. Biochemistry (Mosc); 2011 [cited 2025
Nov 21]; 76: 24-35.
https://doi.org/10.1134/S0006297911010056 |
| 53 | Squillaro T, Alessio N, Di Bernardo G, Özcan S, Peluso G, Galderisi U. Stem Cells and DNA Repair
Capacity: Muse Stem Cells Are Among the Best Performers. Adv Exp Med Biol [Internet]. Springer,
Tokyo; 2018 [cited 2025 Nov 21]; 1103: 103-13.
https://doi.org/10.1007/978-4-431-56847-6_5 |
| 54 | Vargas J, Feltes BC, De Faria Poloni J, Lenz G, Bonatto D. Senescence; An endogenous anticancer
mechanism. Frontiers in Bioscience [Internet]. Bioscience Research Institute; 2011 [cited 2025 Nov
21]; 17: 2616-43.
https://doi.org/10.2741/4074 |
| 55 | Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest [Internet].
American Society for Clinical Investigation; 2018 [cited 2025 Nov 23]; 128: 1238.
https://doi.org/10.1172/JCI95148 |
| 56 | Cianflone E, Torella M, Biamonte F, De Angelis A, Urbanek K, Costanzo FS, Rota M, Ellison-Hughes
GM, Torella D. Targeting Cardiac Stem Cell Senescence to Treat Cardiac Aging and Disease. Cells
[Internet]. MDPI; 2020 [cited 2025 Nov 23]; 9: 1558.
https://doi.org/10.3390/cells9061558 |
| 57 | Kumari R, Jat P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated
Secretory Phenotype. Front Cell Dev Biol [Internet]. Frontiers Media S.A.; 2021 [cited 2025 Nov 23];
9: 645593.
https://doi.org/10.3389/fcell.2021.645593 |
| 58 | Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from
mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol [Internet]. Nature Research; 2020
[cited 2025 Nov 23]; 22: 75.
https://doi.org/10.1038/s41580-020-00314-w |
| 59 | Oguma Y, Alessio N, Aprile D, Dezawa M, Peluso G, Di Bernardo G, Galderisi U. Meta-analysis of
senescent cell secretomes to identify common and specific features of the different senescent
phenotypes: a tool for developing new senotherapeutics. Cell Commun Signal [Internet]. BioMed
Central Ltd; 2023 [cited 2025 Nov 23]; 21: 262.
https://doi.org/10.1186/s12964-023-01280-4 |
| 60 | Beck J, Horikawa I, Harris C. Cellular Senescence: Mechanisms, Morphology, and Mouse Models. Vet
Pathol [Internet]. SAGE Publications Inc.; 2020 [cited 2025 Nov 21]; 57: 747-57.
https://doi.org/10.1177/0300985820943841 |
| 61 | Zhu H, Chan ASL, Narita M. The rise of RAS: how gradual oncogene activation shapes the OIS
spectrum. Genes Dev [Internet]. Cold Spring Harbor Laboratory Press; 2025 [cited 2025 Nov 24]; 39:
936.
https://doi.org/10.1101/gad.352761.125 |
| 62 | Zhu H, Blake S, Kusuma FK, Pearson RB, Kang J, Chan KT. Oncogene-induced senescence: From biology
to therapy. Mech Ageing Dev [Internet]. Elsevier; 2020 [cited 2025 Nov 24]; 187: 111229.
https://doi.org/10.1016/j.mad.2020.111229 |
| 63 | Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garré M,
Giovanni Nuciforo P, Bensimon A, Maestro R, Giuseppe Pelicci P, D'Adda Di Fagagna F.
Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006
444:7119 [Internet]. Nature Publishing Group; 2006 [cited 2025 Nov 24]; 444: 638-42.
https://doi.org/10.1038/nature05327 |
| 64 | Prasanna PGS, Aryankalayil M, Citrin DE, Coleman CN. Radiation-induced pulmonary fibrosis: roles
of therapy-induced senescence and microRNAs. Int J Radiat Biol [Internet]. Taylor and Francis Ltd.;
2023 [cited 2025 Nov 24]; 99: 1027-36.
https://doi.org/10.1080/09553002.2023.2177768 |
| 65 | Saleh T, Bloukh S, Hasan M, Al Shboul S. Therapy-induced senescence as a component of tumor
biology: Evidence from clinical cancer. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer
[Internet]. Elsevier; 2023 [cited 2025 Nov 24]; 1878: 188994.
https://doi.org/10.1016/j.bbcan.2023.188994 |
| 66 | Liao C, Xiao Y, Liu L. The Dynamic Process and Its Dual Effects on Tumors of Therapy-Induced
Senescence. Cancer Manag Res [Internet]. Dove Medical Press Ltd; 2020 [cited 2025 Nov 24]; 12:
13553.
https://doi.org/10.2147/CMAR.S285083 |
| 67 | Litwiniec A, Gackowska L, Helmin-Basa A, Żuryń A, Grzanka A. Low-dose etoposide-treatment induces
endoreplication and cell death accompanied by cytoskeletal alterations in A549 cells: Does the
response involve senescence? The possible role of vimentin. Cancer Cell Int [Internet]. Cancer Cell
Int; 2013 [cited 2025 Nov 23]; 13: 9.
https://doi.org/10.1186/1475-2867-13-9 |
| 68 | Bacca L, Brandariz J, Zacchi F, Zivi A, Mateo J, Labbé DP. Therapy-induced senescence in prostate
cancer: mechanisms, therapeutic strategies, and clinical implications. Gene [Internet]. Elsevier;
2025 [cited 2025 Nov 24]; 972: 149774.
https://doi.org/10.1016/j.gene.2025.149774 |
| 69 | Faheem MM, Seligson ND, Ahmad SM, Rasool RU, Gandhi SG, Bhagat M, Goswami A. Convergence of
therapy-induced senescence (TIS) and EMT in multistep carcinogenesis: current opinions and emerging
perspectives. Cell Death Discov [Internet]. Springer Nature; 2020 [cited 2025 Nov 23]; 6: 51.
https://doi.org/10.1038/s41420-020-0286-z |
| 70 | Liu Y, Lomeli I, Kron SJ. Therapy-Induced Cellular Senescence: Potentiating Tumor Elimination or
Driving Cancer Resistance and Recurrence? Cells [Internet]. Multidisciplinary Digital Publishing
Institute (MDPI); 2024 [cited 2025 Nov 24]; 13: 1281.
https://doi.org/10.3390/cells13151281 |
| 71 | Sikora E, Czarnecka-Herok J, Bojko A, Sunderland P. Therapy-induced polyploidization and
senescence: Coincidence or interconnection? Semin Cancer Biol [Internet]. Academic Press; 2022
[cited 2025 Nov 24]; 81: 83-95.
https://doi.org/10.1016/j.semcancer.2020.11.015 |
| 72 | Chen J, Niu N, Zhang J, Qi L, Shen W, Donkena KV, Feng Z, Liu J. Polyploid Giant Cancer Cells
(PGCCs): The Evil Roots of Cancer. Curr Cancer Drug Targets [Internet]. Bentham Science Publishers
Ltd.; 2019 [cited 2025 Nov 24]; 19: 360-7.
https://doi.org/10.2174/1568009618666180703154233 |
| 73 | Song Y, Zhao Y, Deng Z, Zhao R, Huang Q. Stress-Induced Polyploid Giant Cancer Cells: Unique Way
of Formation and Non-Negligible Characteristics. Front Oncol [Internet]. Frontiers Media S.A.; 2021
[cited 2025 Nov 24]; 11: 724781.
https://doi.org/10.3389/fonc.2021.724781 |
| 74 | Gentric G, Desdouets C. Polyploidization in liver tissue. American Journal of Pathology
[Internet]. Elsevier; 2014 [cited 2025 Nov 24]; 184: 322-31.
https://doi.org/10.1016/j.ajpath.2013.06.035 |
| 75 | Mosieniak G, Sikora E. Polyploidy: the link between senescence and cancer. Curr Pharm Des
[Internet]. Curr Pharm Des; 2010 [cited 2025 Nov 25]; 16: 734-40.
https://doi.org/10.2174/138161210790883714 |
| 76 | Pienta KJ, Hammarlund EU, Austin RH, Axelrod R, Brown JS, Amend SR. Cancer cells employ an
evolutionarily conserved polyploidization program to resist therapy. Semin Cancer Biol [Internet].
Academic Press; 2022 [cited 2025 Nov 24]; 81: 145-59.
https://doi.org/10.1016/j.semcancer.2020.11.016 |
| 77 | Pienta KJ, Hammarlund EU, Axelrod R, Brown JS, Amend SR. Poly-aneuploid cancer cells promote
evolvability, generating lethal cancer. Evol Appl [Internet]. Wiley-Blackwell; 2020 [cited 2025 Nov
25]; 13: 1626-34.
https://doi.org/10.1111/eva.12929 |
| 78 | Kim JY, Choi H, Kim HJ, Jee Y, Noh M, Lee MO. Polyploidization of Hepatocytes: Insights into the
Pathogenesis of Liver Diseases. Biomol Ther (Seoul) [Internet]. Korean Society of Applied
Pharmacology; 2022 [cited 2025 Nov 25]; 30: 391.
https://doi.org/10.4062/biomolther.2022.070 |
| 79 | Zhou X, Zhou M, Zheng M, Tian S, Yang X, Ning Y, Li Y, Zhang S. Polyploid giant cancer cells and
cancer progression. Front Cell Dev Biol [Internet]. Frontiers Media S.A.; 2022 [cited 2025 Nov 25];
10: 1017588.
https://doi.org/10.3389/fcell.2022.1017588 |
| 80 | Liu P, Wang L, Yu H. Polyploid giant cancer cells: origin, possible pathways of formation,
characteristics, and mechanisms of regulation. Front Cell Dev Biol [Internet]. Frontiers Media SA;
2024 [cited 2025 Nov 24]; 12: 1410637.
https://doi.org/10.3389/fcell.2024.1410637 |
| 81 | Hass R, von der Ohe J, Dittmar T. Hybrid Formation and Fusion of Cancer Cells In vitro and In
vivo. Cancers (Basel) [Internet]. MDPI; 2021 [cited 2025 Nov 25]; 13: 4496.
https://doi.org/10.3390/cancers13174496 |
| 82 | Sabe H, Yahara Y, Ishii M. Cell fusion dynamics: mechanisms of multinucleation in osteoclasts and
macrophages. Inflammation and Regeneration 2024 44:1 [Internet]. BioMed Central; 2024 [cited 2025
Nov 25]; 44: 49-.
https://doi.org/10.1186/s41232-024-00360-3 |
| 83 | Zhou X, Platt JL. Molecular and cellular mechanisms of mammalian cell fusion. Adv Exp Med Biol.
2011; 713: 33-64.
https://doi.org/10.1007/978-94-007-0763-4_4 |
| 84 | Zhang J, Qiao Q, Xu H, Zhou R, Liu X. Human cell polyploidization: The good and the evil. Semin
Cancer Biol [Internet]. Academic Press; 2022 [cited 2025 Nov 25]; 81: 54-63.
https://doi.org/10.1016/j.semcancer.2021.04.005 |
| 85 | You B, Xia T, Gu M, Zhang Z, Zhang Q, Shen J, Fan Y, Yao H, Pan S, Lu Y, Cheng T, Yang Z, He X, et
al. AMPK-mTOR-Mediated Activation of Autophagy Promotes Formation of Dormant Polyploid Giant Cancer
Cells. Cancer Res [Internet]. Cancer Res; 2022 [cited 2025 Nov 25]; 82: 846-58.
https://doi.org/10.1158/0008-5472.CAN-21-2342 |
| 86 | Mirzayans R, Murray D. Intratumor Heterogeneity and Treatment Resistance of Solid Tumors with a
Focus on Polyploid/Senescent Giant Cancer Cells (PGCCs). Int J Mol Sci [Internet]. Multidisciplinary
Digital Publishing Institute (MDPI); 2023 [cited 2025 Nov 25]; 24: 11534.
https://doi.org/10.3390/ijms241411534 |
| 87 | Temaj G, Saha S, Chichiarelli S, Telkoparan-Akillilar P, Nuhii N, Hadziselimovic R, Saso L.
Polyploid giant cancer cells: Underlying mechanisms, signaling pathways, and therapeutic strategies.
Crit Rev Oncol Hematol [Internet]. Elsevier; 2025 [cited 2025 Nov 25]; 213: 104802.
https://doi.org/10.1016/j.critrevonc.2025.104802 |
| 88 | Shabo I, Svanvik J, Lindström A, Lechertier T, Trabulo S, Hulit J, Sparey T, Pawelek J. Roles of
cell fusion, hybridization and polyploid cell formation in cancer metastasis. World J Clin Oncol
[Internet]. Baishideng Publishing Group Inc.; 2020 [cited 2025 Nov 25]; 11: 121.
https://doi.org/10.5306/wjco.v11.i3.121 |
| 89 | Pacifico F, Magni F, Leonardi A, Crescenzi E. Therapy-Induced Senescence: Novel Approaches for
Markers Identification. Int J Mol Sci [Internet]. Multidisciplinary Digital Publishing Institute
(MDPI); 2024 [cited 2025 Nov 24]; 25: 8448.
https://doi.org/10.3390/ijms25158448 |
| 90 | Øvrebø JI, Edgar BA. Polyploidy in tissue homeostasis and regeneration. Development [Internet].
Company of Biologists Ltd; 2018 [cited 2025 Nov 25]; 145: dev156034.
https://doi.org/10.1242/dev.156034 |
| 91 | Fujikawa-Yamamoto K, Miyagoshi M, Yamagishi H. Cell cycle, morphology and pluripotency of
octaploid embryonic stem cells in comparison with those of tetraploid and diploid cells. Hum Cell.
2009; 22: 64-71.
https://doi.org/10.1111/j.1749-0774.2009.00070.x |
| 92 | Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular Senescence: Aging,
Cancer, and Injury. https://doi.org/101152/physrev000202018 [Internet]. American Physiological
Society Bethesda, MD ; 2019 [cited 2025 Nov 12]; 99: 1047-78.
https://doi.org/10.1152/physrev.00020.2018 |
| 93 | Zhang W, Chen Y, Li M, Cao S, Wang N, Zhang Y, Wang Y. A PDA-Functionalized 3D Lung Scaffold
Bioplatform to Construct Complicated Breast Tumor Microenvironment for Anticancer Drug Screening and
Immunotherapy. Advanced Science. John Wiley and Sons Inc; 2023; 10.
https://doi.org/10.1002/advs.202302855 |
| 94 | Patra S, Naik PP, Mahapatra KK, Alotaibi MR, Patil S, Patro BS, Sethi G, Efferth T, Bhutia SK.
Recent advancement of autophagy in polyploid giant cancer cells and its interconnection with
senescence and stemness for therapeutic opportunities. Cancer Lett [Internet]. Elsevier; 2024 [cited
2025 Nov 25]; 590: 216843.
https://doi.org/10.1016/j.canlet.2024.216843 |
| 95 | Haidar Ahmad S, El Baba R, Herbein G. Polyploid giant cancer cells, cytokines and cytomegalovirus
in breast cancer progression. Cancer Cell Int. BioMed Central Ltd; 2023; 23.
https://doi.org/10.1186/s12935-023-02971-1 |
| 96 | Ma Y, Shih C-H, Cheng J, Chen H-C, Wang L-J, Tan Y, Zhang Y, Brown DD, Oesterreich S, Lee A V,
Chiu Y-C, Chen Y-C. High-Throughput Empirical and Virtual Screening To Discover Novel Inhibitors of
Polyploid Giant Cancer Cells in Breast Cancer. Anal Chem [Internet]. American Chemical Society; 2025
[cited 2025 Nov 27]; 97: 5498-506.
https://doi.org/10.1021/acs.analchem.4c05138 |
| 97 | Thakur D, Sengupta D, Kar S, Chakrabarti J, Sen S, Hajra S, Laha A, Mahapatra E, Das S, Karmakar
P, Mukherjee S. Aurora Kinase A inhibitor alisertib failed to exert its efficacy on TNBC cells due
to consequential enrichment of polyploid giant cancer cells (PGCCs). Discover oncology [Internet].
Discov Oncol; 2025 [cited 2025 Nov 14]; 16: 2043.
https://doi.org/10.1007/s12672-025-03825-0 |
| 98 | Yazdi N, Pourjamal N, Katainen R, Väänänen J, Dai J, Vähärautio A, Isola J, Kempas M, Le Joncour
V, Laakkonen P, Joensuu H, Barok M. Drug-tolerant persisting polyploid giant cancer cells mediate
resistance to HER2-targeting antibody-drug conjugates. Cancer Lett. Elsevier Ireland Ltd; 2025; 630.
https://doi.org/10.1016/j.canlet.2025.217900 |
| 99 | Go RE, Seong SM, Choi Y, Choi KC. A Fungicide, Fludioxonil, Formed the Polyploid Giant Cancer
Cells and Induced Metastasis and Stemness in MDA-MB-231 Triple-Negative Breast Cancer Cells. Int J
Mol Sci. Multidisciplinary Digital Publishing Institute (MDPI); 2024; 25.
https://doi.org/10.3390/ijms25169024 |
| 100 | Prasanna PG, Citrin DE, Hildesheim J, Ahmed MM, Venkatachalam S, Riscuta G, Xi D, Zheng G, Deursen
J van, Goronzy J, Kron SJ, Anscher MS, Sharpless NE, et al. Therapy-Induced Senescence:
Opportunities to Improve Anticancer Therapy. JNCI Journal of the National Cancer Institute
[Internet]. Oxford University Press; 2021 [cited 2025 Nov 14]; 113: 1285.
https://doi.org/10.1093/jnci/djab064 |
| 101 | Lai P, Liu L, Bancaro N, Troiani M, ..., Alimonti A. Mitochondrial DNA released by senescent tumor
cells enhances PMN-MDSC-driven immunosuppression through the cGAS-STING pathway [Internet].
Immunity. 2025 [cited 2025 Nov 14]. p. 811-25.
https://doi.org/10.1016/j.immuni.2025.03.005 |
| 102 | Cao L, Li K, Li Q, Tong Q, Wang Y, Huang L. The controversial role of senescence-associated
secretory phenotype (SASP) in cancer therapy. Mol Cancer [Internet]. 2025 [cited 2025 Nov 14]; 24:
283.
https://doi.org/10.1186/s12943-025-02475-8 |
| 103 | Xiao S, Qin D, Hou X, Tian L, Yu Y, Zhang R, Lyu H, Guo D, Chen XZ, Zhou C, Tang J. Cellular
senescence: a double-edged sword in cancer therapy. Front Oncol [Internet]. Frontiers Media SA; 2023
[cited 2025 Nov 14]; 13: 1189015.
https://doi.org/10.3389/fonc.2023.1189015 |
| 104 | Kallenbach J, Atri Roozbahani G, Heidari Horestani M, Baniahmad A. Distinct mechanisms mediating
therapy-induced cellular senescence in prostate cancer. Cell Biosci [Internet]. BioMed Central Ltd;
2022 [cited 2025 Nov 14]; 12: 200.
https://doi.org/10.1186/s13578-022-00941-0 |
| 105 | Saleh T, Tyutyunyk-Massey L, Murray GF, Alotaibi MR, Kawale AS, Elsayed Z, Henderson SC, Yakovlev
V, Elmore LW, Toor A, Harada H, Reed J, Landry JW, et al. Tumor cell escape from therapy-induced
senescence. Biochem Pharmacol [Internet]. Elsevier; 2019 [cited 2025 Nov 14]; 162: 202-12.
https://doi.org/10.1016/j.bcp.2018.12.013 |
| 106 | Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW,
Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, et al. A complex secretory program
orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol [Internet]. 2013
[cited 2025 Nov 14]; 15: 978.
https://doi.org/10.1038/ncb2784 |
| 107 | Song KX, Wang JX, Huang D. Therapy-induced senescent tumor cells in cancer relapse. Journal of the
National Cancer Center [Internet]. Chinese National Cancer Center; 2023 [cited 2025 Nov 14]; 3: 273.
https://doi.org/10.1016/j.jncc.2023.09.001 |
| 108 | Rossi M, Abdelmohsen K. The Emergence of Senescent Surface Biomarkers as Senotherapeutic Targets.
Cells [Internet]. Cells; 2021 [cited 2025 Nov 27]; 10.
https://doi.org/10.3390/cells10071740 |
| 109 | Saleh T, Alhesa A, Al-Balas M, Abuelaish O, Mansour A, Awad H, El-Sadoni M, Carpenter VJ, Azab B.
Expression of therapy-induced senescence markers in breast cancer samples upon incomplete response
to neoadjuvant chemotherapy. Biosci Rep [Internet]. Portland Press Ltd; 2021 [cited 2025 Nov 14];
41: BSR20210079.
https://doi.org/10.1042/BSR20210079 |
| 110 | Chakrabarty A, Chakraborty S, Bhattacharya R, Chowdhury G. Senescence-Induced Chemoresistance in
Triple Negative Breast Cancer and Evolution-Based Treatment Strategies. Front Oncol [Internet].
Frontiers Media S.A.; 2021 [cited 2025 Nov 14]; 11: 674354.
https://doi.org/10.3389/fonc.2021.674354 |
| 111 | Hwang HJ, Kang D, Shin J, Jung J, Ko S, Jung KH, Hong SS, Park JE, Oh MJ, An HJ, Yang WH, Ko YG,
Cha JH, et al. Therapy-induced senescent cancer cells contribute to cancer progression by promoting
ribophorin 1-dependent PD-L1 upregulation. Nature Communications 2025 16:1 [Internet]. Nature
Publishing Group; 2025 [cited 2025 Nov 14]; 16: 353-.
https://doi.org/10.1038/s41467-024-54132-1 |
| 112 | Tóth F, Moftakhar Z, Sotgia F, Lisanti MP. In vitro Investigation of Therapy-Induced Senescence
and Senescence Escape in Breast Cancer Cells Using Novel Flow Cytometry-Based Methods. Cells
[Internet]. Multidisciplinary Digital Publishing Institute (MDPI); 2024 [cited 2025 Nov 14]; 13:
841.
https://doi.org/10.3390/cells13100841 |
| 113 | Kim M, Go J, Kwon JH, Jin HJ, Bae YK, Kim EY, Chang EJ, Choi SJ, Kim SW. CD26 Inhibition
Potentiates the Therapeutic Effects of Human Umbilical Cord Blood-Derived Mesenchymal Stem Cells by
Delaying Cellular Senescence. Front Cell Dev Biol [Internet]. Frontiers Media S.A.; 2022 [cited 2025
Nov 14]; 9: 803645.
https://doi.org/10.3389/fcell.2021.803645 |
| 114 | Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends Cell Biol
[Internet]. Elsevier Current Trends; 2018 [cited 2025 Nov 14]; 28: 436-53.
https://doi.org/10.1016/j.tcb.2018.02.001 |
| 115 | Campisi J. Aging, Cellular Senescence, and Cancer. Annu Rev Physiol [Internet]. 2012 [cited 2025
Nov 14]; 75: 685.
https://doi.org/10.1146/annurev-physiol-030212-183653 |
| 116 | Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, Holtz A, Shah S, Sharma V, Ferrucci L,
Campisi J, Schilling B. A proteomic atlas of senescence-associated secretomes for aging biomarker
development. PLoS Biol [Internet]. Public Library of Science; 2020 [cited 2025 Nov 14]; 18:
e3000599.
https://doi.org/10.1371/journal.pbio.3000599 |
| 117 | Feng T, Xie F, Lee LMY, Lin Z, Tu Y, Lyu Y, Yu P, Wu J, Chen B, Zhang G, Tse GMK, To KF, Kang W.
Cellular senescence in cancer: from mechanism paradoxes to precision therapeutics. Mol Cancer
[Internet]. BioMed Central Ltd; 2025 [cited 2025 Nov 14]; 24: 213.
https://doi.org/10.1186/s12943-025-02419-2 |
| 118 | Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM,
Alston S, Academia EC, Kilmarx S, et al. Cellular senescence promotes adverse effects of
chemotherapy and cancer relapse. Cancer Discov [Internet]. American Association for Cancer Research
Inc.; 2016 [cited 2025 Nov 14]; 7: 165.
|
| 119 | Niu N, Mercado-Uribe I, Liu J. Dedifferentiation into blastomere-like cancer stem cells via
formation of polyploid giant cancer cells. Oncogene 2017 36:34 [Internet]. Nature Publishing Group;
2017 [cited 2025 Nov 14]; 36: 4887-900.
https://doi.org/10.1038/onc.2017.72 |
| 120 | Huang P, Wu R, Yang Z, Li Y, Fei F, Yu Y. Polyploid giant cancer cells and tumor budding:
translation from basic research to clinical application. Front Oncol. Frontiers Media SA; 2025; 15:
1611920.
https://doi.org/10.3389/fonc.2025.1611920 |
| 121 | Ahmad SH, El Baba R, Herbein G. Polyploid giant cancer cells, cytokines and cytomegalovirus in
breast cancer progression. Cancer Cell Int [Internet]. BioMed Central Ltd; 2023 [cited 2025 Nov 14];
23: 119.
https://doi.org/10.1186/s12935-023-02971-1 |
| 122 | Zhang Z, Feng X, Deng Z, Cheng J, Wang Y, Zhao M, Zhao Y, He S, Huang Q. Irradiation‐induced
polyploid giant cancer cells are involved in tumor cell repopulation via neosis. Mol Oncol
[Internet]. John Wiley and Sons Ltd; 2021 [cited 2025 Nov 14]; 15: 2219.
https://doi.org/10.1002/1878-0261.12913 |
| 123 | Zhang S, Mercado-Uribe I, Xing Z, Sun B, Kuang J, Liu J. Generation of Cancer Stem-like Cells
through Formation of Polyploid Giant Cancer Cells. Oncogene [Internet]. Nature Publishing Group;
2013 [cited 2025 Nov 14]; 33: 10.1038/onc.2013.96.
https://doi.org/10.1038/onc.2013.96 |
| 124 | Lam Y, Gu J, Yin P. Cellular senescence in cancer: Unveiling dual roles, tumor microenvironment
dynamics and therapeutic innovations (Review). Oncol Lett [Internet]. Spandidos Publications; 2025
[cited 2025 Nov 14]; 30.
https://doi.org/10.3892/ol.2025.15338 |
| 125 | Gao Y, Zuo L, Zheng Y, Sun K, Gao Y, Xu X, Li Z, Zuo D. NOTCH1 inhibition enhances immunogenicity
and sensitizes triple-negative breast cancer to immune checkpoint inhibitors. Breast Cancer Res
[Internet]. Breast Cancer Res; 2025 [cited 2025 Nov 15]; 27.
https://doi.org/10.1186/s13058-025-02045-2 |
| 126 | Chen X, Li Y, Zhou Z, Zhang Y, Chang L, Gao X, Li Q, Luo H, Westover KD, Zhu J, Wei X. Dynamic
ultrasound molecular-targeted imaging of senescence in evaluation of lapatinib resistance in
HER2-positive breast cancer. Cancer Med. John Wiley and Sons Inc; 2023; 12: 19904-20.
https://doi.org/10.1002/cam4.6607 |
| 127 | Xie J, Shu X, Xie Z, Tang J, Wang G. Pharmacological modulation of cellular senescence:
Implications for breast cancer progression and therapeutic strategies. Eur J Pharmacol [Internet].
Elsevier; 2025 [cited 2025 Nov 13]; 997: 177475.
https://doi.org/10.1016/j.ejphar.2025.177475 |
| 128 | Lagoumtzi SM, Chondrogianni N. Senolytics and senomorphics: Natural and synthetic therapeutics in
the treatment of aging and chronic diseases. Free Radic Biol Med [Internet]. Elsevier Inc.; 2021
[cited 2025 Nov 27]; 171: 169-90.
https://doi.org/10.1016/j.freeradbiomed.2021.05.003 |
| 129 | Almudimeegh S, Almutairi MM, Softah A, Alhazzani K, Binobaid L, Alshammari M, Sobeai HMA, Saleh T,
Alotaibi MR, Alhoshani A. Talazoparib and radiation enhance the senolytic efficacy of venetoclax in
therapy-induced senescent triple-negative breast cancer cells. Saudi Pharm J [Internet]. Saudi Pharm
J; 2025 [cited 2025 Nov 13]; 33.
https://doi.org/10.1007/s44446-025-00034-2 |
| 130 | Softah A, Alotaibi MR, Alhoshani AR, Saleh T, Alhazzani K, Almutairi MM, AlRowis R, Alshehri S,
Albekairy NA, Harada H, Boyd R, Chakraborty E, Gewirtz DA, et al. The Combination of Radiation with
PARP Inhibition Enhances Senescence and Sensitivity to the Senolytic, Navitoclax, in Triple Negative
Breast Tumor Cells. Biomedicines [Internet]. Multidisciplinary Digital Publishing Institute (MDPI);
2023 [cited 2025 Nov 13]; 11: 3066.
https://doi.org/10.3390/biomedicines11113066 |
| 131 | Lewińska A, Przybylski P, Adamczyk-Grochala J, Błoniarz D, Litwinienko G, Wnuk M. Senolysis-Based
Elimination of Chemotherapy-Induced Senescent Breast Cancer Cells by Quercetin Derivative with
Blocked Hydroxy Groups. Cancers (Basel) [Internet]. MDPI; 2022 [cited 2025 Nov 15]; 14: 605.
https://doi.org/10.3390/cancers14030605 |
| 132 | Rzeszutek I, Cybularczyk-Cecotka M, Deręgowska A, Stec P, Wnuk M, Kołodziej O, Kałafut J,
Wawruszak A, Witkowski W, Litwinienko G, Lewińska A. New Mitochondria-Targeted Fisetin Derivative
Compromises Mitophagy and Limits Survival of Drug-Induced Senescent Breast Cancer Cells. J Med Chem
[Internet]. American Chemical Society; 2024 [cited 2025 Nov 27]; 67: 17676-89.
https://doi.org/10.1021/acs.jmedchem.4c01664 |
| 133 | Mongiardi MP, Pellegrini M, Pallini R, Levi A, Falchetti ML. Cancer Response to Therapy-Induced
Senescence: A Matter of Dose and Timing. Cancers (Basel) [Internet]. MDPI AG; 2021 [cited 2025 Nov
27]; 13: 484.
https://doi.org/10.3390/cancers13030484 |
| 134 | Bharadwaj D, Mandal M. Senescence in polyploid giant cancer cells: A road that leads to
chemoresistance. Cytokine Growth Factor Rev [Internet]. Pergamon; 2020 [cited 2025 Nov 27]; 52:
68-75.
https://doi.org/10.1016/j.cytogfr.2019.11.002 |
| 135 | Czarnecka-Herok J, Sliwinska MA, Herok M, Targonska A, Strzeszewska-Potyrala A, Bojko A, Wolny A,
Mosieniak G, Sikora E. Therapy-Induced Senescent/Polyploid Cancer Cells Undergo Atypical Divisions
Associated with Altered Expression of Meiosis, Spermatogenesis and EMT Genes. Int J Mol Sci
[Internet]. Int J Mol Sci; 2022 [cited 2025 Nov 27]; 23.
https://doi.org/10.3390/ijms23158288 |
| 136 | Fei F, Zhang D, Yang Z, Wang S, Wang X, Wu Z, Wu Q, Zhang S. The number of polyploid giant cancer
cells and epithelial-mesenchymal transition-related proteins are associated with invasion and
metastasis in human breast cancer. Journal of Experimental and Clinical Cancer Research. BioMed
Central Ltd; 2015; 34.
https://doi.org/10.1186/s13046-015-0277-8 |
| 137 | Salmina K, Vainshelbaum NM, Kreishmane M, Inashkina I, Cragg MS, Pjanova D, Erenpreisa J. The Role
of Mitotic Slippage in Creating a "Female Pregnancy-like System" in a Single Polyploid Giant Cancer
Cell. Int J Mol Sci. Multidisciplinary Digital Publishing Institute (MDPI); 2023; 24.
https://doi.org/10.3390/ijms24043237 |
| 138 | Kudo R, Safonov A, Jones C, Moiso E, Dry JR, Shao H, Nag S, da Silva EM, Yildirim SY, Li Q,
O'Connell E, Patel P, Will M, et al. Long-term breast cancer response to CDK4/6 inhibition defined
by TP53-mediated geroconversion. Cancer Cell. Cell Press; 2024; 42: 1919-1935.e9.
https://doi.org/10.1016/j.ccell.2024.09.009 |
| 139 | Choudhury D, Ghosh D, Mondal M, Singha D, Pothuraju R, Malakar P. Polyploidy and mTOR signaling: a
possible molecular link. Cell Communication and Signaling 2024 22:1 [Internet]. BioMed Central; 2024
[cited 2025 Nov 27]; 22: 196-.
https://doi.org/10.1186/s12964-024-01526-9 |
| 140 | Xuan B, Ghosh D, Cheney EM, Clifton EM, Dawson MR. Dysregulation in Actin Cytoskeletal
Organization Drives Increased Stiffness and Migratory Persistence in Polyploidal Giant Cancer Cells.
Sci Rep [Internet]. Nature Publishing Group; 2018 [cited 2025 Nov 27]; 8: 11935.
https://doi.org/10.1038/s41598-018-29817-5 |
| 141 | Conway PJ, De La Peña Avalos B, Dao J, Montagnino S, Kovalskyy D, Dray E, Mahadevan D. Aurkin-A, a
TPX2-Aurora A small molecule inhibitor disrupts Alisertib-induced polyploidy in aggressive diffuse
large B cell lymphoma. Neoplasia [Internet]. Elsevier; 2024 [cited 2025 Nov 27]; 55: 101014.
https://doi.org/10.1016/j.neo.2024.101014 |
| 142 | Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nature Cell Biology 2019 21:1
[Internet]. Nature Publishing Group; 2019 [cited 2025 Nov 27]; 21: 94-101.
https://doi.org/10.1038/s41556-018-0249-2 |
| 143 | Rodier F, Coppé JP, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR,
Campisi J. Persistent DNA damage signaling triggers senescence-associated inflammatory
|