Original Article - DOI:10.33594/000000831
Accepted 1 November 2025 - Published online
19 November 2025
Na⁺-ATPase, (Na⁺+K⁺)-ATPase and NaPi-II Crosstalk in Ovarian Cancer Cells: Potential Role in Proliferation and Survival
bDepartment of Pharmaceutical Sciences, Health Sciences Center, Federal University of Espírito Santo, Vitória, Espírito Santo, Brazil,
cBiotechnology Program/RENORBIO, Health Sciences Center, Federal University of Espírito Santo, Vitória, Espírito Santo, Brazil,
dBiochemistry Program, Health Sciences Center, Federal University of Espírito Santo, Vitória, Espírito Santo, Brazil
Keywords
Abstract
Background/Aims:
Epithelial ovarian cancer (EOC) is the second leading cause of death among malignant neoplasms of the female reproductive system. In the U.S., approximately 20,890 new EOC cases and 12,730 EOC-related deaths are projected for 2025. Currently, there is no sufficiently accurate screening test for the early detection of EOC, nor have any specific or sensitive EOC markers proven effective in identifying the disease at its initial and potentially curable stages.Methods:
EOC cell lines (A2780, ACRP, ES-2) and the non-tumoral IOSE cell line were analyzed using functional assays, including ATPase activity and phosphate uptake measurements, western blotting, and assays for proliferation, apoptosis, and motility. Pharmacological inhibitors were employed to assess the involvement of the PI3K/Akt/mTOR and MEK/ERK signaling pathways in the regulation of these transporters.Results:
NaPi-IIb and Na⁺-ATPase hyperactivity were associated with increased proliferation, whereas only Na⁺-ATPase supported the survival and motility of EOC cells. In contrast, (Na⁺+K⁺)-ATPase activity was not significantly implicated in EOC progression. Importantly, the PI3K/Akt/mTOR and MEK/ERK pathways modulated these effects in a lineage-dependent manner.Conclusion:
The functional cooperation between NaPi-IIb and Na⁺-ATPase, regulated by major oncogenic pathways, contributes to proliferation, survival, and motility in EOC. These findings reveal a mechanism underlying ovarian cancer progression and highlight NaPi-IIb and Na⁺-ATPase as promising therapeutic targets for the development of more effective and selective treatments.Introduction
Epithelial ovarian cancer (EOC) is the second most common cause of death among neoplasms of the female reproductive system [1]. In the U.S., an estimated 20, 890 new EOC cases and 12, 730 EOC-related deaths are expected in 2025 [2]. Currently, there is no sufficiently accurate screening test for the early detection of EOC, nor have any specific or sensitive EOC markers proven effective in identifying the disease at its initial and potentially curable stages. Consequently, approximately 70% of EOC cases are diagnosed at advanced stages, when metastatic tumor cells have acquired a drug-resistant phenotype, and the 5-year survival rate drops to about 25% (compared with 80% for localized disease) [3]. SLC34A2, which encodes NaPi-IIb, has been shown to be overexpressed across all EOC histological subtypes compared with normal ovarian surface epithelium; therefore, NaPi-IIb has emerged as a promising candidate biomarker for EOC [4, 5].
Despite substantial evidence supporting the clinical significance of targeting NaPi-IIb to combat EOC (for example, using the MX35 antibody) [5, 6,7, 8,9, 10], its regulation and function in ovarian cells remain unclear. One proposed role of NaPi-IIb in the ovary is the maintenance of folate levels essential for oocyte maturation, a theory supported by the expression of NaPi-IIb in the epithelial cells lining the fallopian tubes [11]. NaPi-IIb is a secondary active transporter that utilizes the sodium electrochemical gradient to import phosphate from the extracellular space into the cell cytoplasm. In relation to the establishment and maintenance of this sodium electrochemical gradient in epithelial cells by primary active transporters, the classical ouabain-sensitive (Na⁺+K⁺)-ATPase and a second Na⁺-ATPase—insensitive to ouabain, sensitive to furosemide, and previously cloned in enterocytes—are critical. Notably, the expression of Na⁺-ATPase in ovarian cells has not yet been investigated [12].
This study aimed to investigate the expression of NaPi-IIb and its correlation with the two sodium pumps in EOC cells. Additionally, the potential roles of these transporters in EOC were examined. Herein, we present novel findings indicating that Na⁺-ATPase and NaPi-IIb, but not (Na⁺+K⁺)-ATPase, are modulated during EOC progression. Furthermore, Na⁺-ATPase appears to coordinate with NaPi-II to enhance cellular proliferation in EOC. In contrast, only Na⁺-ATPase promotes the survival and motility of EOC cells.
Materials and Methods
Reagents
ATP (sodium salt), ouabain, furosemide, phosphonoformic acid (PFA), sodium chloride, potassium chloride,
magnesium chloride, EDTA, EGTA, Hepes, Tris, sodium deoxycholate and sodium orthovanadate were purchased
from
Sigma-Aldrich. Sodium pyrophosphate was purchased from Reagen. SDS was purchased from GE Healthcare.
Sucrose was
from Merck. Wortmannin was purchased from Calbiochem. U0126, polyclonal phospho-PKB (Ser-473), polyclonal
PKB,
polyclonal phosphor-ERK (Thr-202/Tyr-204), polyclonal ERK, polyclonal GAPDH and polyclonal β-actin
antibodies
were from Cell Signaling Technology. Anti-NPT-2b was from Santa Cruz Biotechnology. 32Pi was obtained from
the
Brazilian Institute of Energetic and Nuclear Research (São Paulo, Brazil). γ-32Pi-ATP was synthesized
according
to the procedures described by Maia et al. [13]. All other reagents were of the highest purity available.
Cell culture
Human EOC cells A2780, ACRP and ES-2 were kindly provided by Dr. Patrice J. Morin,
NIH, USA, and maintained in RPMI 1640 medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum
(FBS,
Gibco/Invitrogen) and stabilized solution of penicillin (100 un/mL), streptomycin (100 µg/mL) and
amphotericin B
(250 µg/mL) (Gibco/Invitrogen). Immortalized cell line IOSE was kindly provided by Dr. Nelly Auersperg of
the
Canadian Ovarian Tissue Bank (University of British Columbia, Vancouver, Canada), and cultured in 1:1
mixture of
medium 199 and MCDB 105 (Sigma-Aldrich), supplemented with 10% FBS, L-Glutamine (5 mM), and stabilized
solution
of penicillin (100 un/ml), streptomycin (100 ug/ml) and amphotericin B (250 ug/ml). Cells were grown at
37°C in
a humidified atmosphere of 95% air and 5% CO2. Cells were preincubated with different compounds indicated
in the
in serum-depleted medium. A2780 is an ovarian carcinoma cell line sensitive to cisplatin, established from
tumor
tissue from an untreated patient. ACRP is a cell line originated from the intermittent treatment of A2780
with
cisplatin, and it was determined that the cisplatin IC50 for A2780 and ACRP was 0.75 uM and 3 uM,
respectively
[14]. ES-2 is a clear cell ovarian carcinoma (CCOC) that exhibits low to moderate resistance to various
chemotherapeutic agents such as doxorubicin, cisplatin, carmistine and etoposide. IOSE is an immortalized
cell
line with epithelial ovarian cell characteristics without tumorigenic function in Nu/nu mice [15].
Antibodies
All primary antibodies were rabbit IgG and purchased from Cell Signaling Technology®. Primary antibodies
were
used at a 1:1000 dilution and incubated overnight. The phosphorylated secondary antibodies were applied at
a
1:4000 dilution, while the total ones were used at a 1:5000 dilution. All secondary antibodies were
incubated
for 1 hour.
Measurement of (Na++K+)-ATPase and Na+-ATPase activities
ATPase activity was measured according to the method described by Grubmeyer and Penefsky [16]. Briefly,
treated
cells were washed in PBS, harvested, and cleared by centrifugation for 5 min at 600 x g, and incubated for
30
min in lysis buffer (1 mM EGTA, 0.1% sodium deoxycholate, 20 mM Hepes (pH 7.0), 250 mM sucrose). The
composition
of the standard assay medium was 10 mM MgCl2, 8.3 mM ATP (specific activity 90mCi/mmol [γ-32P] ATP), 33 mM
HEPES
(pH 7.0), 120 mM NaCl, and 50 mM KCl. The reaction was started by the addition of cell lysate. After 10
min, the
reaction was stopped with charcoal activated with 0.1 N HCl. The 32Pi released to the medium was measured
in
liquid scintillation counter (Packard Tri-Carb 2100 TR). (Na++K+)-ATPase activity was determined by its
sensitivity to 1 mM ouabain and expressed in nanomoles of Pi per milligram of protein per minute.
Na+-ATPase
activity was evaluated in the presence of 1 mM ouabain and in the absence of KCl added to the medium and
determined by its sensitivity to 2 mM furosemide and expressed in nanomoles of Pi per milligram of protein
per
minute. Protein quantification was determined by the Folin phenol method, using BSA as standard.
Measurement of sodium-dependent phosphate uptake in EOC cells
Sodium-dependent phosphate uptake was measured by the uptake of radiolabeled phosphate into EOC cell
monolayers.
Cells were pretreated with the indicated conditions in serum-free RPMI medium. Measurement of phosphate
uptake
was initiated by the addition of transport medium containing NaCl 140 mM, KCl 5 mM, MgCl2 1 mM, CaCl2 1.5
mM,
HEPES 15 mM (pH 7.4), Na2HPO4/NaH2PO4.H2O 0.1 mM (pH 7.4) and 32Pi (1 mCi/mmole), which allowed the
analysis of
the total phosphate transport. Another transport buffer containing choline chloride instead of NaCl was
used to
measure Na+-independent Pi uptake. Uptake was stopped after 10 min by aspiration of the transport medium
and
washing the cells with ice-cold PBS. Cell monolayers were solubilized by the addition of 1% SDS and the
radioactivity was counted in liquid scintillation counter (Packard Tri-Carb 2100 TR). Absolute values of
phosphate uptake were calculated. Na+-dependent Pi transport was calculated by subtraction of the
Na+-independent component from the total Pi uptake in the presence of Na+ and expressed in nanomoles of Pi
per
milligram of protein per minute. Protein quantification was determined by the Folin phenol method, using
BSA as
standard.
Western Blot
The cells were washed with PBS 1x, harvested, and incubated for 40 min in lysis buffer (25 mM Tris (pH
7.5), 50
mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 10 mM SDS, 10 mM NaF, 5 mM sodium vanadate,
5 mM
sodium pyrophosphate, protease inhibitor mixture (Sigma-Aldrich) and cleared by centrifugation for 10 min
at 15,
000 x g. The supernatant was retained, and the protein concentrations were determined by the Folin phenol
method, using BSA as standard. Proteins were resolved in SDS-polyacrylamide gels and transferred to
polyvinylidene fluoride membranes (Millipore), according to the manufacturer’s instructions. After
antibody
labeling, detection was acquired with ECL-prime (GE Amersham Biosciences).
Phagokinetic Track Motility Assay
The phagokinetic track motility assay is based on the ability of a moving cell to clear gold particles
from its
path to create a measurable track on a colloidal gold-coated plate [17]. Briefly, 24-well plates were
coated
with the colloidal gold particles. Cells were trypsinized and resuspended in culture medium at a density
of 2 x
103 cells/ml, and 0.5 milliliter of the cell suspension, treated or not, were added to each gold
particle-coated
well. After incubation at 37°C in 5% CO2 for 16 h, the tracks created by single migrating cells were
visible and
photographs under 10X magnification were taken using a Leica inverted phase-contrast microscope (Leica DM
IL
LED, Leica Microsystems) equipped with Quick Imaging system. Cell motility was evaluated by measuring the
areas
free of the gold particles using the ImageJ software (version 1.6.0). At least 20 cells per condition were
analyzed in three independent experiments.
CFSE assay
Cell proliferation was analyzed using a carboxyfluorescein succinimidyl ester (CFSE) kit (CellTraceTM CFSE
Cell
Proliferation Kit, Invitrogen) according to the manufacturer's instructions. Briefly, cells were stained
with
CFSE and seeded in 12-wells plates for 24 hours. Following, cells from some wells were harvested with
trypsin-EDTA (0.25 mg/ml trypsin and 1 mg/ml EDTA), centrifuged at 2000 x g for 5 minutes, resuspended in
culture medium and analyzed by flow cytometry (BD FACScan) to evaluate the binding intensity with CFSE to
the
cells at the beginning of each described treatment. The remaining cells were treated with furosemide (2.0
mM) or
vehicle (0.4% NaOH) or PFA (1.0 mM) or only with culture medium and cultured for 24 hours. After 24 hours
of
treatment, cells were harvested with trypsin-EDTA, centrifuged, and resuspended in culture medium, and
then
analyzed on a flow cytometer. The results were analyzed by FlowJo software (version 7.6.2) and expressed
as a
percentage of divided cells. The experiments were performed in triplicate.
Apoptosis assay
For determination of cell death by apoptosis, 2 x 105 cells were seeded in 12-well culture plates. After
48
hours, cells were treated with furosemide (2.0 mM) or vehicle (0.4% NaOH) or PFA (1.0 mM) or with culture
medium
alone and incubated at 37°C under 5% CO2 for 24 hours. The supernatant was collected (for evaluation of
the
cells in suspension) and the adherent cells were gently collected with PBS-EDTA (0.5 mM). Samples were
then
centrifuged, resuspended in binding buffer (Annexin V-FITC Kit, BD, USA), incubated for 15 minutes in the
dark
at room temperature with Annexin V-FITC, then incubated with propidium iodide and analyzed immediately by
flow
cytometry. Data were analyzed using the FlowJo software (version 7.6.2). The experiments were run in
triplicate.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA).
Results
are presented as the mean ± SEM. For comparisons between two groups, an unpaired Student's t-test was
performed.
Comparisons among three or more groups were conducted using one-way ANOVA followed by Bonferroni post hoc
test,
when applicable. Statistical significance was defined as p < 0.05.
Results
Expression profile of NaPi-IIb in EOC cells
Our data proved that all tested ovarian cancer cells express NaPi-IIb in higher magnitude than
immortalized
non-tumoral normal epithelial ovarian cells (IOSE) cells. Amongst the EOC cells, A2780 and ACRP express
the
transporter two-fold more than ES-2 (p<0.05) (Fig. 1). Altogether, our results suggest that serous
ovarian
carcinoma (SOC) overexpresses NaPi-IIb in a higher magnitude when compared to CCOC. Even though these are
both
low prognostic diseases, mainly due to late diagnosis and chemoresistance, it is worthwhile to mention
that the
later occur much more frequently than the later.
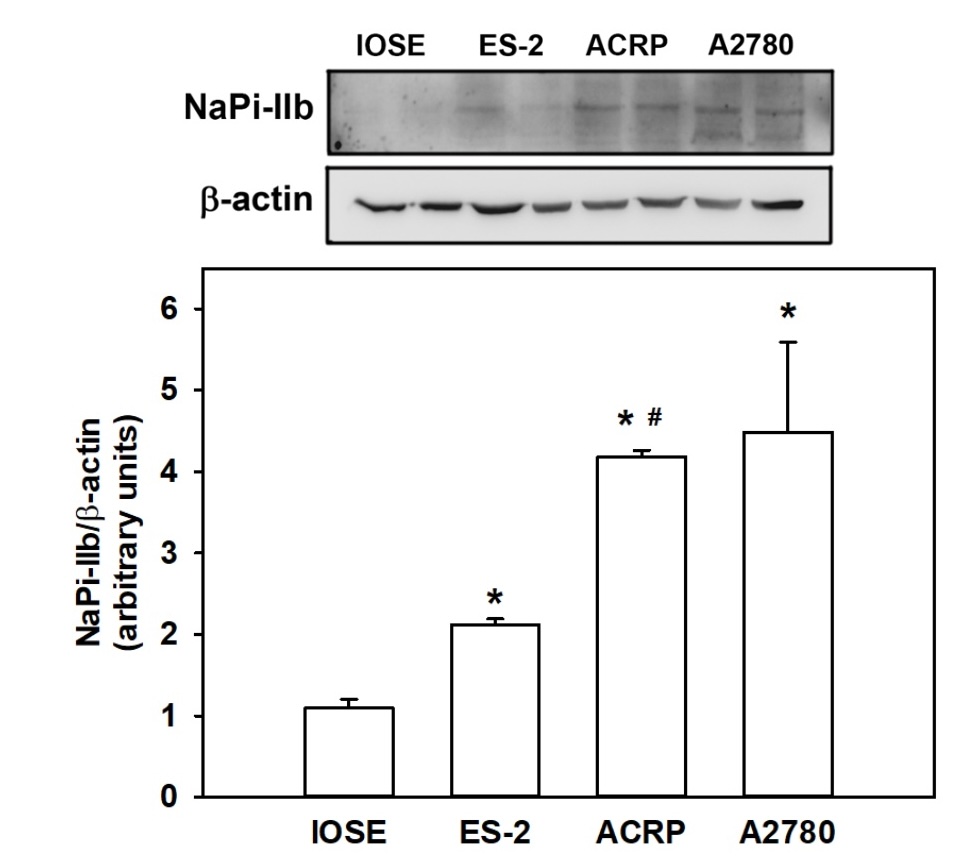
Fig. 1: NaPi-IIb expression was analyzed in EOC cells: A2780 and ACRP, both SOC; ES-2, CCOC; IOSE, immortalized non-tumoral normal epithelial ovarian cells. N=3; *p<0.05 compared to IOSE. #p<0.05 compared to ES-2.
Sodium dependent phosphate uptake in EOC cells
Motivated by the fact that EOC cells overexpress NaPi-IIb compared to the normal counterpart IOSE cells,
we
argued whereas there was transporter activity of sodium dependent phosphate uptake sensitive to
phosphonoformate
acid (PFA), a selective inhibitor of NaPi-II. Sodium dependent phosphate uptake sensitive to PFA increased
in a
dose dependent manner in all lineages studied, as ACRP=A2780>ES-2>IOSE (Fig. 2) (p<0.05). These
findings corroborate to our previous data concerning the expression of the protein NaPi-IIb (Fig. 1). The
kinetic parameters of the sodium dependent phosphate transporter were calculated following the equation
v=Vmax
[S]/K0.5+[S] and are summarized in Table 1. Vmax was higher in ACRP and in A2780, (0.3514 ± 0.0264 and
0.2701 ±
0.0153 nmol Pi x mg-1 x min-1, respectively) than in ES-2 (0.2035 ± 0.0225 nmol Pi x mg-1 x min-1)
(p<0.05).
On the other hand, IOSE had higher Vmax than EOC cells (0.0812 ± 0.0220 nmol Pi x mg-1 x min-1) (p<0,
005).
The differences observed regarding NaPi-II kinetic parameters in ovarian cells lead us to investigate the
regulation and the function of NaPi-II in EOC.
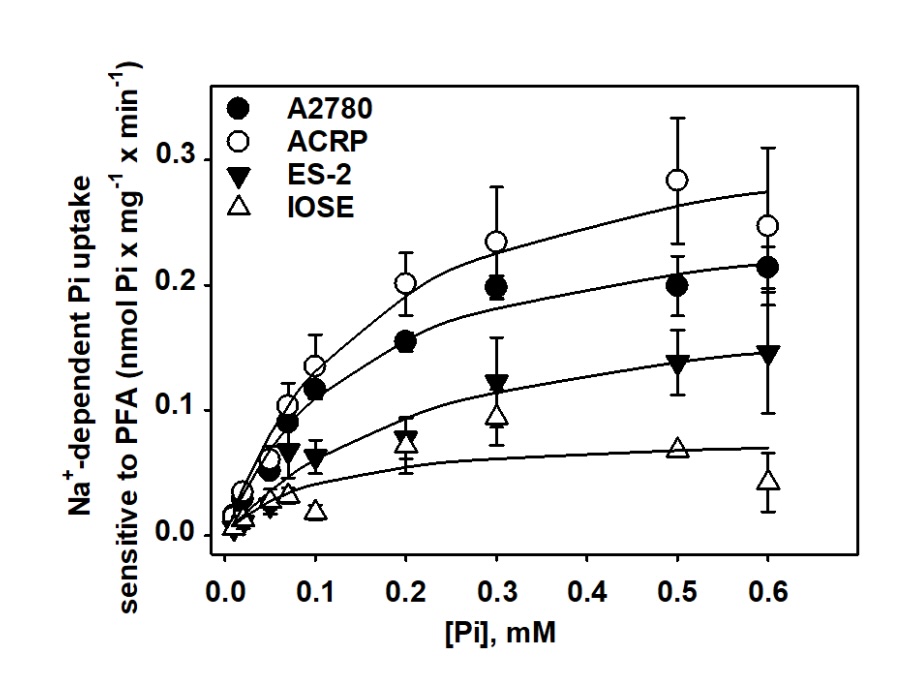
Fig. 2: Sodium dependent phosphate uptake, sensitive to PFA, in ovarian cell lines. The experiment was conducted in a dose dependent manner, with phosphate varying from 0.01 to 0.6 mM 32Pi (1 mCi/mmol Pi) was used as a tracer, and the protein activity expressed in nmol Pi x min-1 x mg-1. N=4.
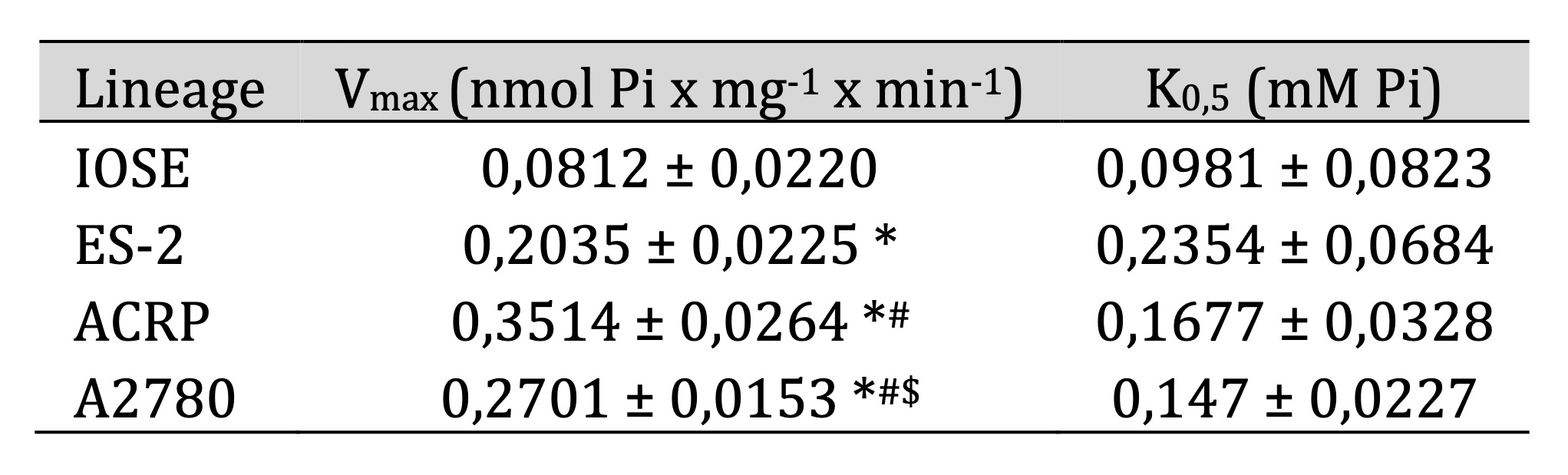
Table 1: Kinetic parameters of the sodium dependent phosphate uptake, sensitive to PFA, in ovarian cells. * p<0,05 compared to IOSE; #p<0,05 in comparison with ES-2; $p<0,05 in relation to ACRP
ATPase and Na+ATPase
NaPi-IIb is a secondary active transporter, thus depends on a primary active transporter, such as
(Na⁺+K⁺)-ATPase, to function properly. As shown in Fig. 3, preliminary results (N=2) suggest that
(Na⁺+K⁺)-ATPase activity may partially modulate NaPi-II activity in the presence of sodium [140 mM] or
choline
[140 mM] in EOC cells. Although statistical analysis could not be performed due to the limited number of
replicates, an approximately 50% decrease in NaPi-II activity was observed following 30 min treatment of
ovarian
cells with ouabain, supporting a potential functional link between these transporters that warrants
further
investigation.
We further questioned the proportional contribution of (Na++K+)-ATPase and
Na+-ATPase on ovarian malignant transformation, and got interest results: i) the ratio of the
activities of (Na++K+)-ATPase and Na+-ATPase was 3.81-, 1.92-, 2.14-, and
1.03-fold in IOSE, ES-2, ACRP, and A2780, respectively (Figure 4) (p<0.05); ii) the activity of
(Na++K+)-ATPase did not influence ovarian carcinogenesis, despite the fact that its
activity was sustained in aggressive tumors, therefore it might contribute to the phenomenon; iii)
Na+-ATPase was over-activated in EOC cells vs. IOSE cells; iv) whereas
(Na++K+)-ATPase was the same in EOC vs. IOSE cells, Na+-ATPase and
NaPi-II
profiles were similar in the ovary. One possibility is that Na+-ATPase might provide the fine
sodium
gradient that is probable required for the higher activity of NaPi-II in EOC, especially in SOC (the most
frequent and highly aggressive form of the disease) (Fig. 5). We then postulated that
Na+-ATPase
would modulate NaPi-II sensitive to PFA. We observed that furosemide inhibited the sodium dependent
phosphate
uptake in a dose dependent manner, the maximum effect being at 2.0 mM. Under this experimental condition,
NaPi-II decreased from 0.264 ± 0.027 without furosemide to 0.135 ± 0.029 nmol Pi x mg-1 x
min-1in the presence of furosemide in A2780; from 0.244 ± 0.027 (in the absence of furosemide)
to
0.121 ± 0.006 nmol Pi x mg-1 x min-1(in the presence of furosemide) in ACRP; from
0.169 ±
0.007 (without furosemide) to 0.091 ± 0.007 nmol Pi x mg-1 x min-1(with furosemide)
(Fig.
5) (p<0.001 for all cases). Taking in conjunction, our results strongly support the correlation between
the
activity of NaPi-II and Na+-ATPase in ovarian cells.
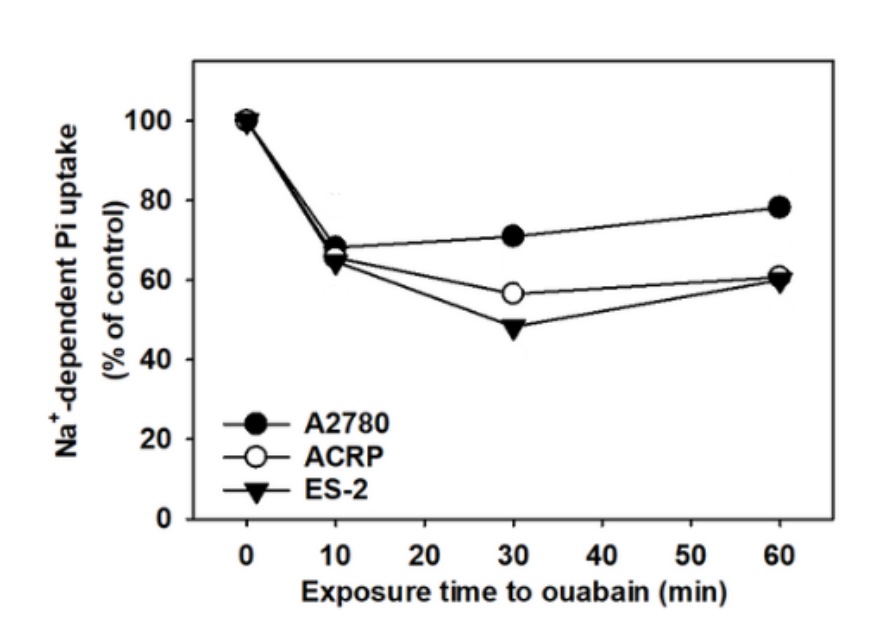
Fig. 3: Sodium dependent phosphate uptake in EOC cells: A2780, ACRP, and ES-2 cells were treated with ouabain 1 mM, and NaPi-II was evaluated in a time-course fashion in these cells.32Pi (1 mCi/mmol Pi) was used as a reaction tracer. Results were expressed as percentage of the sodium phosphate caption. N=2.
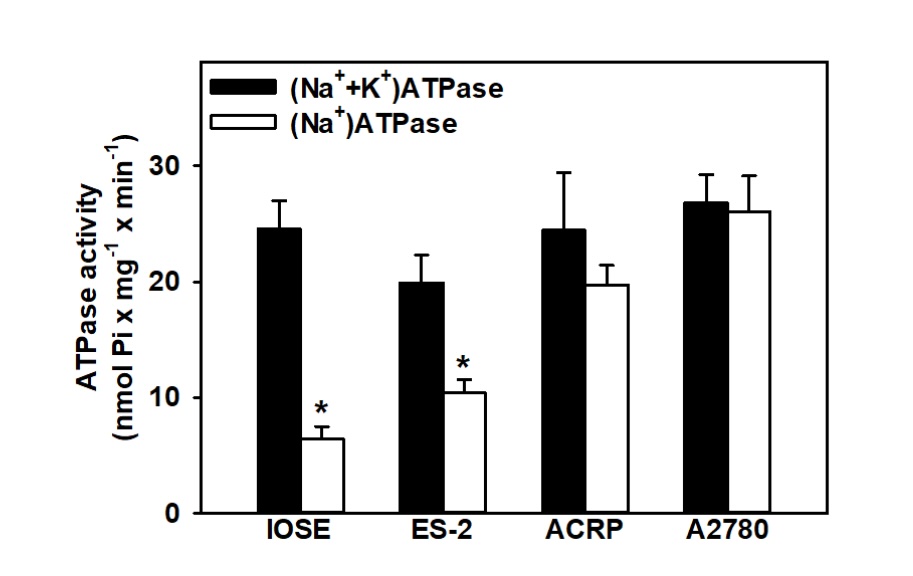
Fig. 4: Correlation between Na+-ATPase and (Na++K+)-ATPase activities in ovarian lineages (IOSE, ES-2, ACRP and A2780). N=4. *p<0.05 in comparison with the (Na++K+)-ATPase activity within the same lineage.
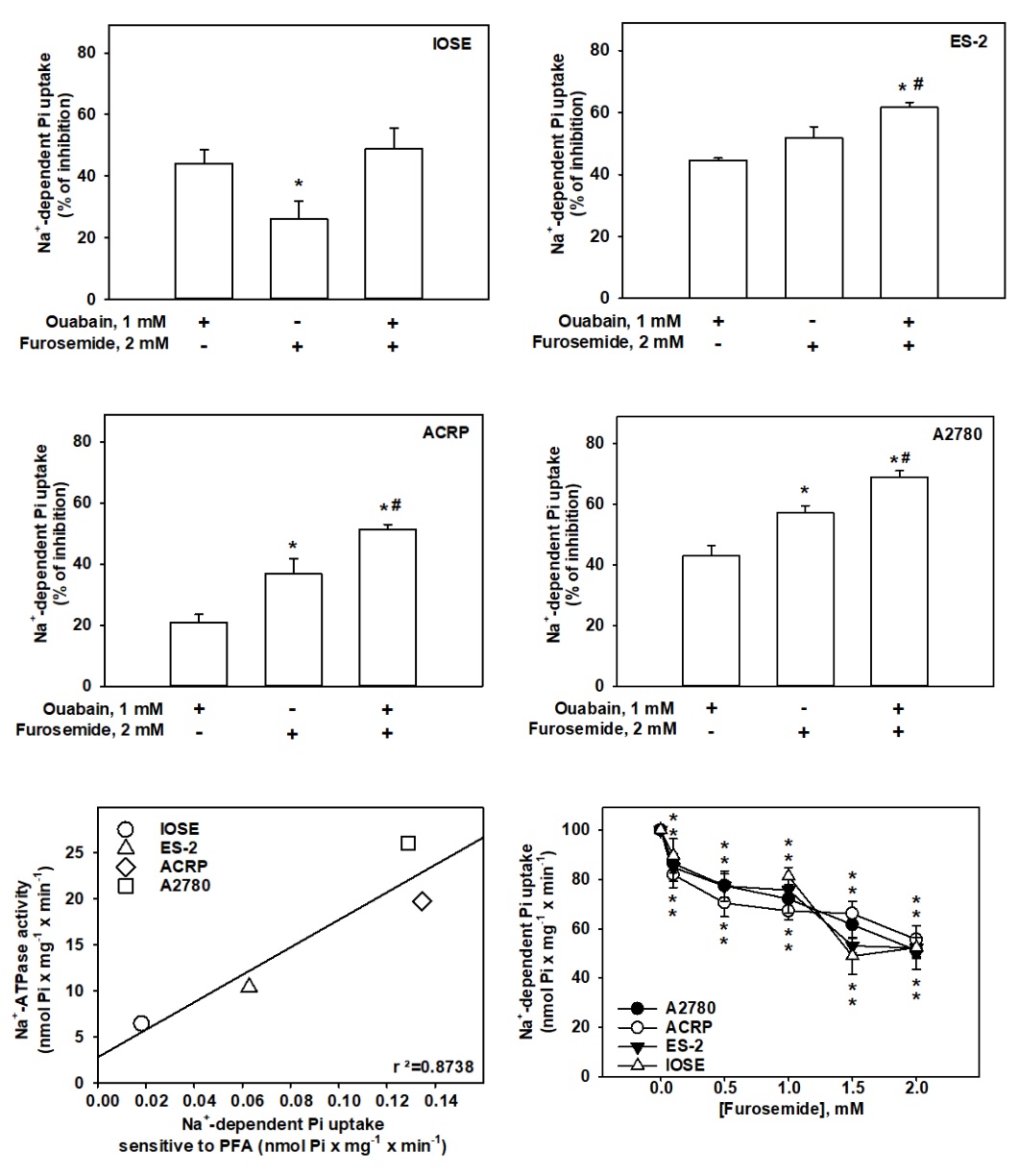
Fig. 5: Modulation of sodium dependent phosphate uptake in ovarian cells modulated by (Na++K+)-ATPase and Na+-ATPase. (A-D) Ouabain 1.0 mM and Furosemide 2.0 mM were added as shown in the Fig..32Pi (1 mCi/mmol Pi) was used as a tracer. Enzyme activity was expressed as percentage in relation to control. N=6. *p< 0,01 in comparison with cells treated with ouabain or furosemide#p<0,05. (E) Fig. shows the correlation between Na+-ATPase and the sodium dependent phosphate uptake, sensitive to PFA, in ovarian cells. N=4. r²=0.8738. (F) The Fig. demonstrates the positive effect of Na+-ATPase on NaPi-II sensitive to PFA.32Pi (1 mCi/mmol Pi) was used as a tracer. Data were expressed as a percentage of control. N=6. *p< 0.05 in comparison with non-treated cells.
Na+-ATPase activity insensitive to ouabain and sensitive to furosemide, and
of NaPi-II sensitive to PFA modulate ovarian cells motility and survival
Cell motility, measured as arbitrary unit, was reduced in 31% (p<0.001), 40% (p<0.0001), 50%
(p<0.0001)
and 35% (p<0.0005) in IOSE, ES-2, ACRP and A2780 by furosemide (2.0 mM), respectively. In turn,
motility was
decreased in IOSE by PFA (1.0 mM) in 17% (p<0.05) (Fig. 6). Our findings suggest that NaPi-II is
unlikely to
regulate the motility of EOC cells, being its effect restricted to IOSE. On the other hand,
Na+-ATPase contributes, at least partially, to EOC cells' motility (Fig. 7), which is a key
phenomenon for cancer cells' metastasis. Next, we argued whether NaPi-II, sensitive to PFA and Na+-ATPase,
sensitive to furosemide and insensitive to ouabain, would play a role in ovarian cell proliferation.
Therefore,
we conducted the carboxyfluorescein succinimidyl ester incorporation assay (CFSE). Cells were labelled
with
CFSE, and then treated with furosemide (2.0 mM), vehicle (NaOH 0.4%) or PFA (1.0 mM). Proliferative cells
were
detected by flow cytometry. Furosemide 2.0 mM reduced the ovarian cellular proliferation by 17%
(p<0.05), 45%
(p<0.001), 28% (p<0.01) and 46% (p<0.001) in IOSE, ES-2, ACRP and A2780, respectively. On the
other
hand, PFA reduced ovarian cell proliferation in 38% (p<0.05), 40% (p<0.001), 55% (p<0.01) and 62%
(p<0.001) in IOSE, ES-2, ACRP and A2780, respectively (Fig. 8). Of note, the proliferation index of
IOSE was
significantly lower than the EOC cells. Therefore, our results suggest that whereas NaPi-II modulates the
proliferative index of normal and cancerous ovarian cells, Na+ATPase induces the proliferation of
cancerous
cells exclusively. In conjunction, our data strongly suggest that whereas NaPi-II regulates the
proliferation of
normal and cancerous ovarian cells, Na+ATPase likely increases the proliferative index of EOC cells more
prominently than normal ovarian cells.
Evaluation of the role of both transporters activity in ovarian cells survival was assessed by the annexin
V/Pi
analysis assay using flow cytometry. Our results show that furosemide (2.0 mM) increased the percentage of
apoptotic cells, 2.4-. 1.8- (p<0.0001); 2.0- (p<0.0001) and 2.0-fold (p<0.0001), compared to
control,
in IOSE (Fig. 9), ES-2 (Fig. 10), ACRP (Fig. 11) e A2780 (Fig. 12), respectively. On the other side, PFA
increased the percentage of apoptotic cells exclusively in ES-2 (1.6-fold; p<0.0001), as shown in Fig.
10.
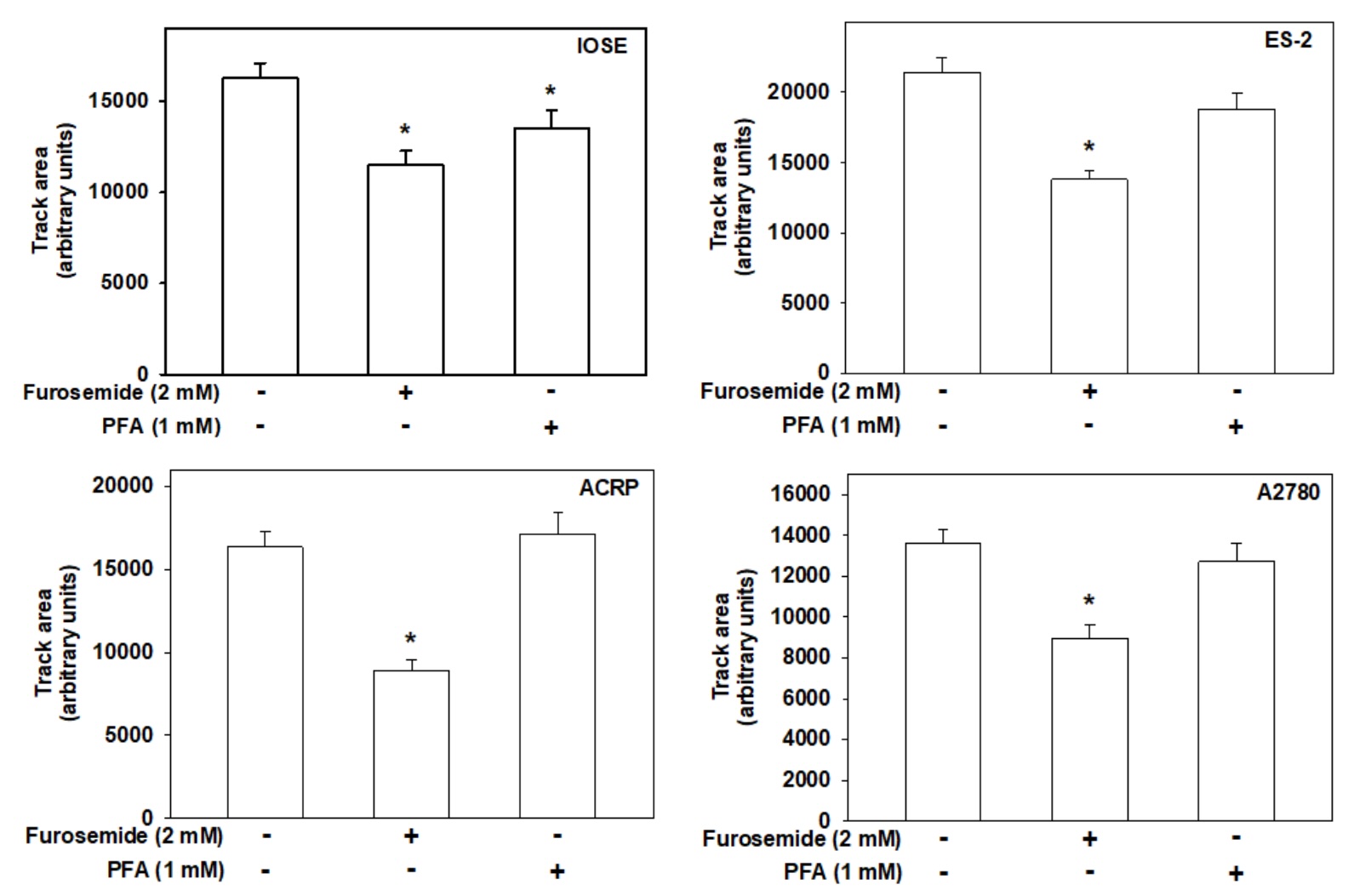
Fig. 6: Colloidal gold assay was used to analyze ovarian cells' motility. Cells were plated into wells covered with a thin gold layer, then treated with furosemide (2.0 mM), vehicle (NaOH 0.4%) or PFA (1.0 mM) for 16 hours. While the furosemide-treated cells decreased the motility of all ovarian cells evaluated, PFA treatment comprised the same effect exclusively in IOSE. *p<0.05 in comparison with non-treated cells.
Fig. 7: Role of NaPi-II and Na+-ATPase in ovarian cells' motility. The colloidal gold motility assay was used to evaluate ovarian cells' motility. Cells were photographed, and the area displaced by the cells was quantified by the Image J Software. N = 3.
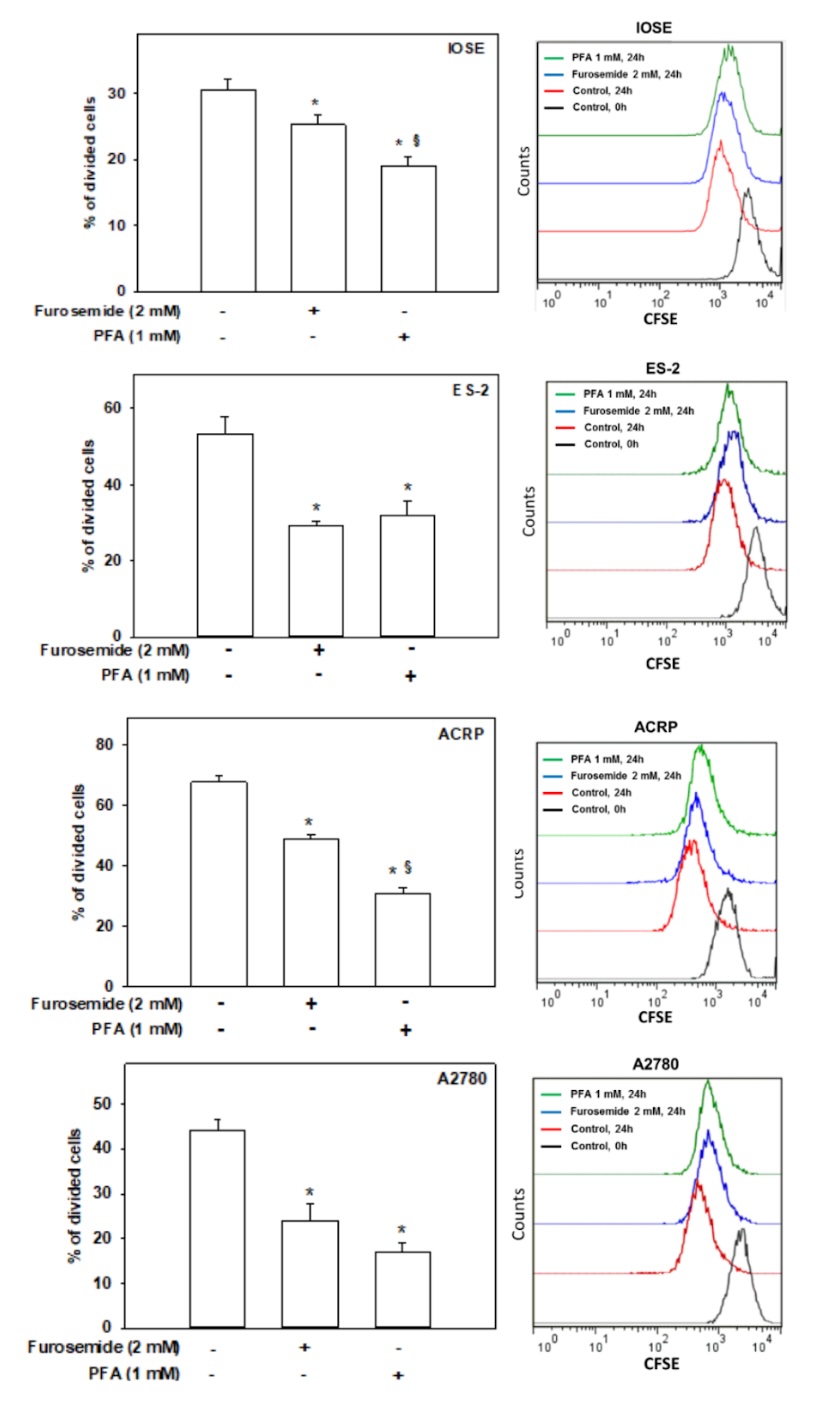
Fig. 8: Effect of NaPi-II and Na⁺-ATPase on ovarian cell proliferation. Cell proliferation was assessed using the carboxyfluorescein succinimidyl ester (CFSE) incorporation assay and analyzed by flow cytometry. The bar graphs represent the mean ± SD of three independent replicates (N=3), while the line graph illustrates a representative CFSE experiment. Cells were treated with furosemide (2.0 mM) or PFA (1.0 mM) to evaluate the roles of Na+-ATPase and NaPi-II in ovarian cells, respectively. *p<0.01 compared to non-treated cells; §p<0.05 compared to cells treated with furosemide.
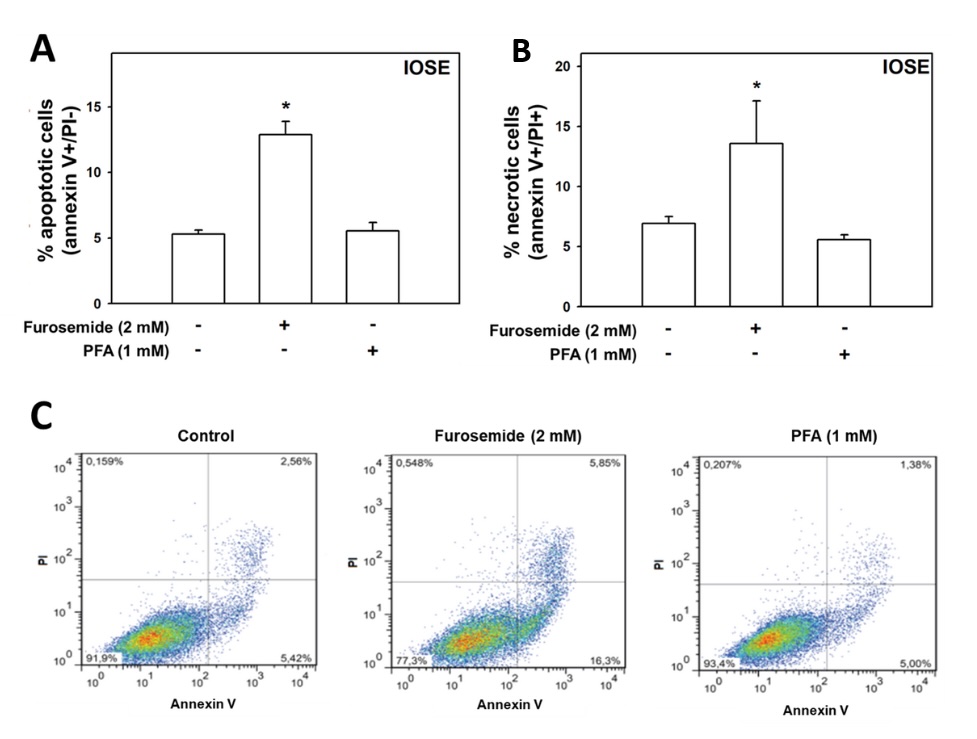
Fig. 9: Evaluation of the role of the NaPi-II cotransporter and Na⁺-ATPase in ovarian epithelial cells' death. Non-cancerous ovarian IOSE cells were seeded and, after 48 hours, treated for 24 hours with furosemide (2,0 mM) or PFA (1,0 mM). Following treatment, cells were stained with annexin V-FITC and propidium iodide (PI) to assess the percentage of apoptotic (annexin V-positive/PI-negative) and necrotic (annexin V-positive/PI-positive) cells. N = 6. *p<0.01 compared to untreated cells.

Fig. 10: Evaluation of the role of the NaPi-II cotransporter and Na⁺-ATPase in ovarian epithelial cells' death. EOC ES-2 cells were seeded and, after 48 hours, treated for 24 hours with furosemide (2,0 mM) or PFA (1,0 mM). Following treatment, cells were stained with annexin V-FITC and propidium iodide (PI) to assess the percentage of apoptotic (annexin V-positive/PI-negative) and necrotic (annexin V-positive/PI-positive) cells. N = 6. *p<0.01 compared to untreated cells.

Fig. 11: Evaluation of the role of the NaPi-II cotransporter and Na⁺-ATPase in ovarian epithelial cells' death. EOC ACRP cells were seeded and, after 48 hours, treated for 24 hours with furosemide (2,0 mM) or PFA (1,0 mM). Following treatment, cells were stained with annexin V-FITC and propidium iodide (PI) to assess the percentage of apoptotic (annexin V-positive/PI-negative) and necrotic (annexin V-positive/PI-positive) cells. N = 6. *p<0.01 compared to untreated cells.

Fig. 12: Evaluation of the role of the NaPi-II cotransporter and Na⁺-ATPase in ovarian epithelial cells' death. EOC A2780 cells were seeded and, after 48 hours, treated for 24 hours with furosemide (2,0 mM) or PFA (1,0 mM). Following treatment, cells were stained with annexin V-FITC and propidium iodide (PI) to assess the percentage of apoptotic (annexin V-positive/PI-negative) and necrotic (annexin V-positive/PI-positive) cells. N = 6. *p<0.01 compared to untreated cells.
MEK/ERK and PI3K/AKT/mTOR signalling pathways regulate NaPi-II in ovarian cells
Considering the key role of MAPK (MEK/ERK) and PI3K/AKT/mTOR signalling pathways in cancer, we decided to
investigate their status in ovarian cells, as well as their role in NaPi-II and Na+-ATPase activity
modulation.
Protein expression was evaluated by Western blot, and data are compiled in Fig. 13. MAPK pathway was
differentially expressed in EOC cells compared to IOSE cells. Indeed, the activation of MAPK signalling
pathway
was more intense in EOC vs. IOSE cells. Amongst EOC cells, however, the observation was
ACRP>A2780>ES-2.
On the other hand, PI3K pathway was activated in ACRP and A2780 cells, which is known to have the protein
constitutively activated, when compared with IOSE and ES-2, as previously observed by Hua et al, 2008 and
2009
[18, 19].
Considering the expression profile of the MAPK and the PI3K pathways in ovarian cells, we sought to
investigate
their effect on NaPi-II activity in the same cell lines studied. To do so, cells were cultured in the
presence
or in the absence of ERK inhibitor, U0126 (1.0 µM) or PI3K inhibitor, wortmannin (10-7M). We concluded
that in
all ovarian cells, NaPi-II activity was stimulated by the PI3K/AKT/mTOR pathway as the co-transporter
activity
decreased to 43% (p<0, 01), 27% (p<0, 01), 67% (p<0, 001) and 40% (p<0, 01) in IOSE, ES-2,
ACRP e
A2780, respectively, in the presence of wortmannin. On the other hand, the MAPK pathway is unlikely to
modulate
NaPi-II activity in all ovarian cells. In fact, U0126 reduced the activity of NaPi-IIb in 46% (p<0,
05), 47%
(p<0, 05) and 36% (p<0, 05) in ES-2, ACRP and A2780, respectively. Nonetheless, no significant
effect was
observed in IOSE cells. These results are shown in Fig. 14.
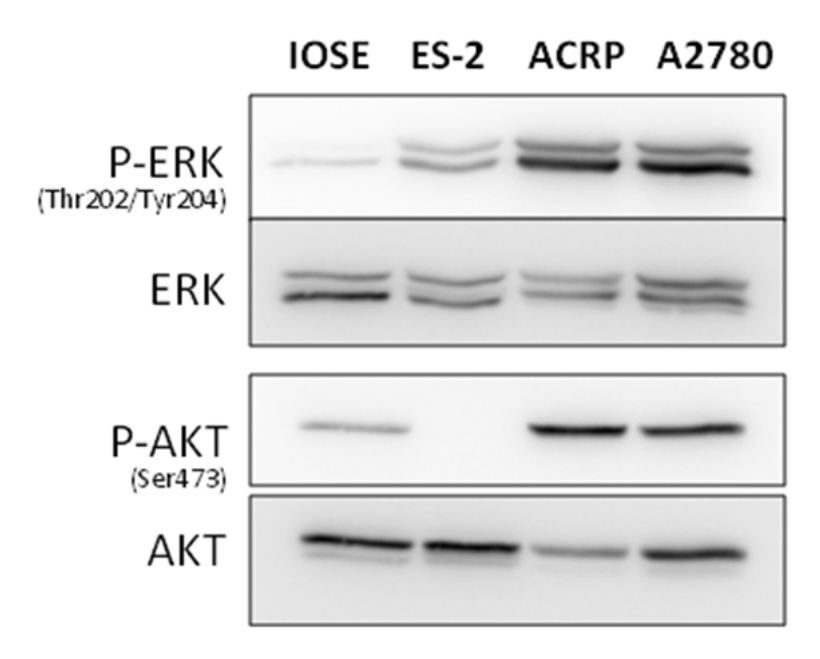
Fig. 13: Profiles of MAPK and PI3K pathways in EOC and IOSE cells by Western blot. N = 3.
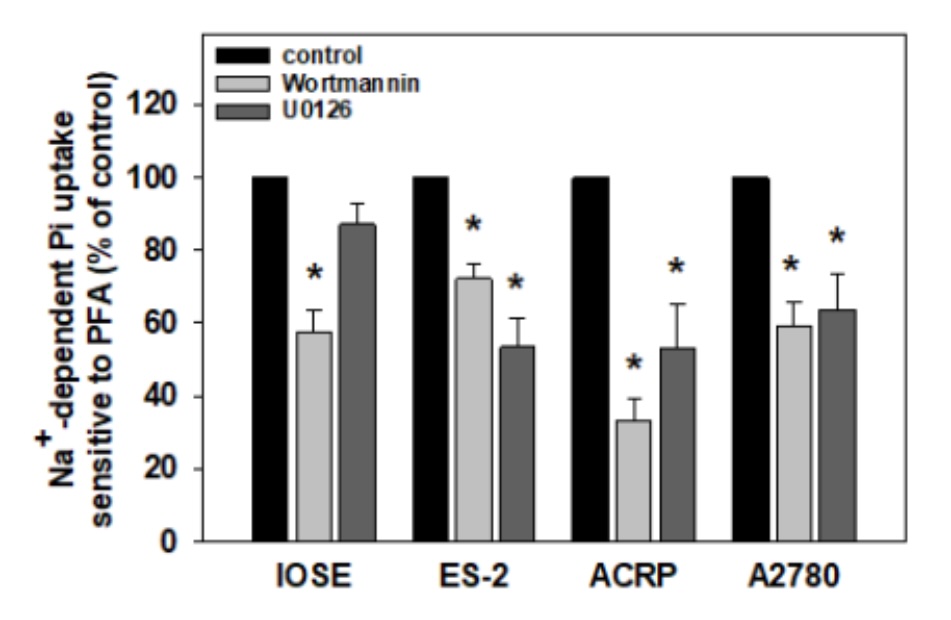
Fig. 14: Modulation of NaPi-II activity by MAPK and PI3K pathways in ovarian cells. Cells were treated for 2 hours with wortmannin (10-7 M) or U0126 (10 uM). Sodium-dependent phosphate uptake sensitive to PFA activity was measured using 32Pi (1 mCi/mmol Pi) as a tracer and was expressed as percentage of control. N=3. * p<0.05 compared to the control.
Discussion
Recent evidence highlights that phosphate metabolism is not merely a secondary biochemical process in malignancies but rather serves as a central regulatory axis essential for sustaining oncogenic growth and metabolic plasticity [20]. Elevated intracellular phosphate levels provide the necessary substrate for ATP synthesis, nucleotide production, and activation of key signalling kinases, including Akt, ERK, and mTOR, thereby supporting increased cell proliferation, biosynthetic activity, and metabolic reprogramming in cancer cells [21]. Moreover, dysregulated phosphate homeostasis has been linked to alterations in tumor immunity, extracellular matrix remodeling, and epithelial–mesenchymal transition (EMT), further underscoring phosphate as a multifaceted determinant of tumor progression and microenvironmental adaptation [22, 23].
Within this broader metabolic context, the NaPi family of sodium-dependent phosphate transporters (SLC34 and SLC20) has emerged as a key regulatory hub in cancer-associated phosphate homeostasis [24]. Among these, NaPi-IIb (SLC34A2) is notable for its pronounced overexpression in various epithelial malignancies, including ovarian, lung, colorectal, and gastric cancers, where it imparts proliferative and survival advantages to tumor cells [24]. In addition to mediating phosphate uptake, NaPi-IIb has attracted growing interest as both a diagnostic and therapeutic target. Several antibody–drug conjugates (ADCs) and monoclonal antibodies targeting NaPi-IIb are currently under investigation, showing promising preclinical efficacy and tumor selectivity with minimal toxicity in non-malignant tissues [25]. Conversely, the phosphate exporter XPR1 (SLC53A1) plays an equally important yet complementary role, regulating intracellular phosphate efflux and influencing tumor aggressiveness and metabolic balance [26]. This reciprocal regulation between NaPi-mediated import and XPR1-mediated export suggests that cancer cells dynamically adjust phosphate fluxes to meet anabolic demands while avoiding cytotoxic phosphate overload. Such a mechanism implies that distinct tumor types may utilize differential phosphate-handling strategies, determined by their metabolic context and signaling architecture [26].
Our present findings are consistent with this model, revealing a functional coupling between NaPi-IIb and the PI3K/Akt and MEK/ERK signaling cascades. These pathways are well established as key regulators of transporter expression and function, integrating nutrient sensing, growth factor responses, and metabolic control [27]. Consistent with previous reports in other tumor systems, our data indicate that NaPi-IIb activation is lineage-dependent and mechanistically linked to oncogenic kinase activity, further reinforcing its role as an adaptive metabolic element within EOC cells [4].
From a translational perspective, these results support the notion that targeting phosphate metabolism represents a promising yet underexplored therapeutic approach in EOC [26]. The tissue-restricted expression profile of NaPi-IIb, combined with its functional interdependence with PI3K and MEK signaling, underscores its potential for tumor-selective intervention [28, 29]. Furthermore, combination strategies utilizing phosphate transporter blockade in conjunction with PI3K or MEK inhibitors may yield synergistic antitumor effects by simultaneously disrupting the metabolic and signaling dependencies that sustain tumor viability and progression.
In this study, we investigated the role of the phosphate co-transporter NaPi-IIb (encoded by the SLC34A2 gene) and the Na⁺-ATPase enzyme in EOC cell lines, as well as their relationship with signaling pathways implicated in tumorigenesis, including PI3K/Akt/mTOR and MEK/ERK. The functional modulation of these proteins was assessed in key cellular processes, such as proliferation, motility, phosphate uptake, and cell viability. Tumor cell lines A2780, ACRP (cisplatin-resistant), and ES-2 (CCOC) were utilized alongside the non-tumorigenic IOSE line to compare tumor and non-tumor expression and functional patterns of the proteins of interest.
Our findings confirm that NaPi-IIb is markedly overexpressed in EOC cell lines compared to the non-tumorigenic IOSE line [30, 31]. Notably, the study by Nurgalieva et al. (2021) further indicates that NaPi-IIb may serve as a potential biomarker in ovarian cancer patients. Its upregulation is functionally associated with a significant increase in phosphate (Pi) uptake, which was effectively inhibited by PFA. The enhanced Pi uptake observed in tumor cells was more pronounced and demonstrated a dose-dependent response to PFA, indicating increased transporter activity in the malignant context. These results strongly support a pivotal role for NaPi-IIb in sustaining the proliferative capacity of EOC cells. In agreement with our observations, Nagayoshi and colleagues (2022) reported that another phosphate transporter, XPR1 (encoded by the SLC53A1 gene), also contributes to the proliferation of CCOC, underscoring the broader relevance of phosphate homeostasis in ovarian cancer biology [32]. Supporting our hypothesis, pharmacological inhibition with PFA significantly reduced (p<0.05) the proliferative index in tumor cell lines, while a milder effect was observed in IOSE cells. This differential sensitivity reinforces the notion that tumor cells are more dependent on elevated Pi influx to support metabolic activity and cell cycle progression than normal cells [29].
Conversely, the effects of PFA on cell motility were limited to IOSE cells, with no notable impact on tumor cells. This finding suggests that, in the tumor context, NaPi-IIb primarily supports proliferation rather than migration, whereas in non-tumorigenic cells, even subtle changes in phosphate transport can influence cellular motility. These results indicate that NaPi-IIb-mediated regulation of motility is not a significant mechanism driving EOC progression. In contrast, Russo and colleagues (2018) demonstrated that, in MDA-MB-231 cells—a widely used in vitro model of triple-negative breast cancer—blockade of phosphate transport with PFA significantly impaired migratory capacity. These findings underscore the importance of phosphate uptake in maintaining pro-tumorigenic features such as motility in this breast cancer model [33].
The functional activity of Na⁺-ATPase was also assessed, revealing that this enzyme is significantly hyperactivated in tumor cell lines compared to IOSE cells. These findings suggest a reprogramming of ion transport mechanisms associated with tumorigenesis, consistent with emerging evidence that ion transporters may play non-canonical roles in cancer progression [34]. Notably, in lung carcinoma, the (Na⁺+K⁺)-ATPase α1 subunit has been shown to cooperate with endogenous ligands to actively reshape the immune microenvironment and promote immune escape [34]. In this context, our data further support the concept that dysregulation of ion transport systems—such as Na⁺-ATPase, the Na⁺/Pi-II cotransporter, and other ATPases—may represent a hallmark of oncogenic reprogramming, with potential implications for tumor cell adaptation, survival, and resistance to therapy.
Na⁺-ATPase activity is insensitive to the classical inhibitor ouabain but responsive to furosemide, suggesting the involvement of atypical ATPases or alternative regulatory mechanisms in ovarian malignant transformation. Supporting our findings, previous studies have shown that furosemide inhibits proliferation of poorly differentiated gastric cancer cells via the Na⁺/K⁺/2Cl⁻ cotransporter, inducing G₀/G₁ cell-cycle arrest and highlighting its ability to impair tumor growth through modulation of sodium transport [35]. Notably, as furosemide treatment significantly reduced sodium-dependent phosphate uptake, cell proliferation, and motility in tumor cells without inducing immediate cell death—thereby excluding nonspecific cytotoxic effects—these results further reinforce the functional relevance of this enzyme in carcinogenesis [36].
Numerous studies have also linked (Na⁺+K⁺)-ATPase to cancer progression: ouabain targeting the α3 isoform of the enzyme inhibits proliferation and induces apoptosis in renal and lung carcinoma models [37], while blockade of (Na⁺+K⁺)-ATPase by cardiac glycosides diminishes proliferation, migration, invasion, and macropinocytosis in breast and colorectal cancer cells [38]. Additionally, acute inhibition disrupts the Na⁺-dependent glycolytic flux in breast cancer, further connecting Na⁺-ATPase function to the metabolic reprogramming characteristic of malignancy [39].
Furthermore, Na⁺-ATPase activity surpassed that of (Na⁺+K⁺)-ATPase in the analyzed cell lines, exerting a stronger influence on proliferation and migration than NaPi-II itself. This suggests that Na⁺-ATPase may play a central role in supporting tumor cell survival and aggressiveness. Taken together, these complementary findings across different sodium transporters and cancer models underscore the critical role of atypical Na⁺ pumps in tumor biology, paving the way for novel therapeutic strategies targeting sodium homeostasis in cancer.
The relationship between NaPi-IIb and (Na⁺+K⁺)-ATPase was also investigated. Although the latter partially modulates the activity of the co-transporter, its activity levels did not correlate with ovarian carcinogenesis, suggesting that its functional contribution, while present, is secondary. In contrast, Na⁺-ATPase significantly modulated both motility and survival of tumor cells, thereby emerging as a more promising potential therapeutic target.
Regarding intracellular signaling, the data indicate that both the PI3K/Akt/mTOR and MEK/ERK pathways contribute to the functional regulation of NaPi-IIb, exhibiting lineage-dependent variations. Basal phosphorylation of Akt at Ser473 was elevated in A2780 and ACRP cells, and inhibition with wortmannin significantly reduced phosphate uptake in these lines, suggesting that the PI3K/Akt pathway is functionally coupled to NaPi-IIb activation. In contrast, this effect was attenuated in ES-2 cells. This pattern suggests that alternative pathways may compensate for phosphate transport regulation in this lineage, or that NaPi-IIb plays a less prominent functional role in ES-2 cells.
Consistent with our observations, other solute carriers—such as the bicarbonate transporter SLC4A7—have also been shown to promote tumor progression by engaging the PI3K/Akt/mTOR signaling cascade, as reported in head and neck squamous cell carcinoma (HNSCC), where SLC4A7 activation enhanced epithelial-to-mesenchymal transition and metastatic potential [40]. Similarly, the involvement of the MEK/ERK axis in modulating ion transport and cellular responses is supported by studies on trigeminal neuralgia, in which modulation of the MAPK pathway altered sodium channel activity and neuronal sensitivity [41]. Collectively, these findings reinforce the concept that ion transporters, beyond their canonical roles in homeostasis, may participate in oncogenic signaling and phenotypic reprogramming via lineage- and context-specific kinase pathways.
Regarding the MEK/ERK pathway, inhibition with U0126 markedly reduced phosphate uptake in cell lines exhibiting high ERK1/2 phosphorylation (A2780 and ACRP), but not in IOSE cells, which displayed low basal pathway activation. These findings suggest that, similar to PI3K/Akt, the MEK/ERK pathway regulates NaPi-IIb in a lineage-specific manner, underscoring the importance of detailed molecular characterization in the design of targeted therapeutic strategies. Consistently, in non-small cell lung cancer (NSCLC), downregulation of SLC34A2 (NaPi-IIb) significantly suppressed cell growth, migration, and invasion, an effect associated with reduced activation of both PI3K/Akt and Ras/Raf/MEK signaling pathways [27].
To assess the role of phosphate in the homeostasis of non-tumorigenic lung cells, Chang and colleagues (2006) reported that high concentrations of phosphate promote increased cell viability and significantly upregulate the expression of phosphate transporters in these cells. Furthermore, they found that elevated phosphate levels stimulate the phosphorylation of Akt and other kinases, including MEK, ERK, and Raf-1. Conversely, treatment of lung cells with PFA—a known phosphate transporter inhibitor—resulted in inhibition of Akt phosphorylation and subsequent reduction in cell growth, suggesting a role for phosphate in Akt-mediated cell survival [37]. Additionally, Jin and colleagues (2009) demonstrated that overexpression of NaPi-IIb led to phosphate-mediated activation of Akt and promoted increased carcinogenesis and proliferation of non-small cell lung carcinoma (NSCLC) cells [42]. This parallel regulation suggests that the functional coupling of NaPi-IIb to MEK/ERK and other pathways is conserved across tumor types, further reinforcing its potential as a context-dependent therapeutic target.
Although our findings strongly support a mechanistic link between NaPi-IIb expression, phosphate uptake, and activation of oncogenic kinases, the directionality of this relationship warrants further investigation. It remains possible that the enhanced proliferative and signaling responses observed following NaPi-IIb upregulation are secondary to increased intracellular phosphate availability, which fuels ATP and nucleotide synthesis and thereby sustains kinase activation [21]. Conversely, transporter-independent functions of NaPi-IIb cannot be excluded. Previous studies have suggested that certain membrane transporters, including members of the SLC34 and SLC20 families, may act as molecular scaffolds or modulators of intracellular signaling independent of their transport activity [24].
Conclusion
Whether NaPi-IIb directly participates in signaling complex formation or indirectly modulates downstream cascades through phosphate flux remains to be determined. Future experiments employing transport-deficient NaPi-IIb mutants or controlled phosphate supplementation and deprivation will be essential to disentangle these possibilities and clarify the precise causal relationship between NaPi-IIb activity and oncogenic signaling in EOC. Nonetheless, the results of this study indicate that hyperactivity of NaPi-IIb and Na⁺-ATPase is associated with proliferation, survival, and motility of EOC cells. The functional modulation of these proteins by pathways such as PI3K/Akt/mTOR and MEK/ERK, in a lineage-dependent manner, highlights promising molecular targets for the development of more effective and selective therapeutic approaches for EOC treatment.
Acknowledgements
The authors would like to thank PhD. Celso Caruso Neves, from the Federal University of Rio de Janeiro, for the technical support and collaboration. Author Contributions: G.M.S. conceived and designed the study, performed the experiments, analyzed the data, and revised the manuscript. T.M.P. analyzed the data, contributed to data interpretation, drafted, wrote and revised the manuscript. B.S.M. contributed to data interpretation. R.P.S.A. contributed to experimental procedures, and data interpretation. M.G.M., S.M.S.B. and J.M.S.P assisted with literature review and reference checking. L.B.A.R. supervised the project, contributed to study design and data interpretation, and critically revised the manuscript. All authors read and approved the final version of the manuscript. Funding: This study received no specific research funding. The authors acknowledge financial support from the Coordination for the Improvement of Higher Education Personnel (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - CAPES, Brazil) through a doctoral fellowship granted to G.M.S. Disclosure of AI Assistance: The authors used ChatGPT (OpenAI, 2025) and Perplexity.AI to assist in checking English grammar and improving language clarity. The authors carefully reviewed and verified all content to ensure accuracy and integrity of the scientific information.
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have no conflicts of interest to declare.
References
| 1 | Sambasivan S. Epithelial ovarian cancer: Review article. Cancer Treat Res Commun. 2022;33:100629.
https://doi.org/10.1016/j.ctarc.2022.100629 |
| 2 | Siegel RL, Kratzer TB, Giaquinto AN, Sung H, Jemal A. Cancer statistics, 2025. CA Cancer J Clin
2025;75(1):10-45.
https://doi.org/10.3322/caac.21871 |
| 3 | Stewart C, Ralyea C, Lockwood S. Ovarian Cancer: An Integrated Review. Semin Oncol Nurs
2019;35(2):151-156.
https://doi.org/10.1016/j.soncn.2019.02.001 |
| 4 | Banerjee S, Drapkin R, Richardson DL, Birrer M. Targeting NaPi2b in ovarian cancer. Cancer Treat
Rev 2023;112:102489.
https://doi.org/10.1016/j.ctrv.2022.102489 |
| 5 | Cerri MF, Rezende LCD, Paes MF, Silva IV, Rangel LBA. The Cotransporter NaPi-IIb: Characteristics,
Regulation and its Role in Carcinogenesis. Appl Cancer Res 2010;30(1):197-203.
|
| 6 | Abane R, Mezger V. Roles of heat shock factors in gametogenesis and development. FEBS J
2010;277(20):4150-4172.
https://doi.org/10.1111/j.1742-4658.2010.07830.x |
| 7 | Bäck T, Andersson H, Divgi CR, Hultborn R, Jensen H, Lindegren S. 211At radioimmunotherapy of
subcutaneous human ovarian cancer xenografts: evaluation of relative biologic effectiveness of an
alpha-emitter in vivo. J Nucl Med 2005;46(15):2061-2067.
|
| 8 | Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev
Cancer 2003;3(7):502-516.
https://doi.org/10.1038/nrc1123 |
| 9 | Finstad CL, Lloyd KO, Federici MG, Divgi C, Venkatraman E, Barakat RR. Distribution of
radiolabeled monoclonal antibody MX35 F(ab')2 in tissue samples by storage phosphor screen image
analysis: evaluation of antibody localization to micrometastatic disease in epithelial ovarian
cancer. Clin Cancer Res 1997;3(8):1433-1442.
|
| 10 | Andersson H, Cederkrantz E, Bäck T, Divgi C, Elgqvist J, Himmelman J. Intraperitoneal
alphaparticle radioimmunotherapy of ovarian cancer patients: pharmacokinetics and dosimetry of
(211)At-MX35 F(ab')2-a phase I study. J Nucl Med 2009;50(7):1153-1160.
https://doi.org/10.2967/jnumed.109.062604 |
| 11 | Kiyamova RG, Vlasenkova RA, Bulatova LF. Sodium-dependent phosphate transporter NaPi2b as a
candidate for targeted therapy: features of structure, function, and expression (In Russ.). Adv Mol
Oncol 2024;11(2):74-84.
https://doi.org/10.17650/2313-805X-2024-11-2-74-84 |
| 12 | Al-Ghoul M, Valdes R Jr. Mammalian cardenolides in cancer prevention and therapeutics. Ther Drug
Monit 2008;30(2):234-328.
https://doi.org/10.1097/FTD.0b013e31816b90ff |
| 13 | Maia JC, Gomes SL, Juliani MH. Preparation of (c-32P)- and (a-32P)-nucleoside triphosphates with
high specific activity, in: Morel CM, ed. Genes and antigens of parasites: a laboratory manual
proceedings. Rio de Janeiro, Fundação Oswaldo Cruz, 1983, pp. 144-157.
|
| 14 | Sherman-Baust CA, Weeraratna AT, Rangel LBA, Pizer ES, Cho KR, Schwartz DR. Remodeling of the
extracellular matrix through overexpression of collagen VI contributes to cisplatin resistance in
ovarian cancer cells. Cancer Cell 2003;3(4):377-386.
https://doi.org/10.1016/S1535-6108(03)00058-8 |
| 15 | Maines-Bandiera SL, Kruk PA, Auersperg N. Simian virus 40-transformed human ovarian surface
epithelial cells escape normal growth controls but retain morphogenetic responses to extracellular
matrix. Am J Obstet Gynecol 1992;167(3):729-735.
https://doi.org/10.1016/S0002-9378(11)91579-8 |
| 16 | Grubmeyer C, Penefsky HS. The presence of two hydrolytic sites on beef heart mitochondrial
adenosine triphosphatase. J Biol Chem 1981;256(8):3718-3727.
https://doi.org/10.1016/S0021-9258(19)69514-1 |
| 17 | Scott WN, McCool K, Nelson J. Improved method for the production of gold colloid monolayers for
use in the phagokinetic track assay for cell motility. Anal Biochem 2000;287(2):343-344.
https://doi.org/10.1006/abio.2000.4866 |
| 18 | Hua K, Feng W, Cao Q, Zhou X, Lu X, Feng Y. Estrogen and progestin regulate metastasis through the
PI3K/AKT pathway in human ovarian cancer. Int J Oncol 2008;33(5):959-967.
|
| 19 | Hua K, Din J, Cao Q, Feng W, Zhang Y, Yao L. Estrogen and progestin regulate HIF-1alpha expression
in ovarian cancer cell lines via the activation of Akt signaling transduction pathway. Oncol Rep
2009;21(4):893-898.
https://doi.org/10.3892/or_00000300 |
| 20 | Schiliro C, Firestein BL. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting
Enhanced Growth and Proliferation. Cells 2021;10(5):1056.
https://doi.org/10.3390/cells10051056 |
| 21 | Arnst JL, Beck GR Jr. Modulating phosphate consumption, a novel therapeutic approach for the
control of cancer cell proliferation and tumorigenesis. Biochem Pharmacol 2021;183:114305.
https://doi.org/10.1016/j.bcp.2020.114305 |
| 22 | Liu X, Zhou X, Xu H, He Z, Shi X, Wu S. SLC34A2 Regulates the Proliferation, Migration, and
Invasion of Human Osteosarcoma Cells Through PTEN/PI3K/AKT Signaling. DNA Cell Biol
2017;36(9):775-780.
https://doi.org/10.1089/dna.2017.3750 |
| 23 | Sun X, Li Z, Wang X, He J, Wu Y. Inorganic Phosphate as "Bioenergetic Messenger" Triggers M2-Type
Macrophage Polarization. Adv Sci (Weinh). 2024;11(13):e2306062.
https://doi.org/10.1002/advs.202306062 |
| 24 | Nurgalieva AK, Popov VE, Skripova VS, Bulatova LF, Savenkova DV, Vlasenkova RA. Sodium-dependent
phosphate transporter NaPi2b as a potential predictive marker for targeted therapy of ovarian
cancer. Biochem Biophys Rep 2021;28:101104.
https://doi.org/10.1016/j.bbrep.2021.101104 |
| 25 | Lin K, Rubinfeld B, Zhang C, Firestein R, Harstad E, Roth L, Tsai SP, Schutten M, Xu K,
Hristopoulos M, Polakis P. Preclinical Development of an Anti-NaPi2b (SLC34A2) Antibody-Drug
Conjugate as a Therapeutic for Non-Small Cell Lung and Ovarian Cancers. Clin Cancer Res. 2015 Nov
15;21(22):5139-50.
https://doi.org/10.1158/1078-0432.CCR-14-3383 |
| 26 | Bondeson DP, Paolella BR, Asfaw A, Rothberg MV, Skipper TA, Langan C, Mesa G, Gonzalez A, Surface
LE, Ito K, Kazachkova M, Colgan WN, Warren A, Dempster JM, Krill-Burger JM, Ericsson M, Tang AA,
Fung I, Chambers ES, Abdusamad M, Dumont N, Doench JG, Piccioni F, Root DE, Boehm J, Hahn WC,
Mannstadt M, McFarland JM, Vazquez F, Golub TR. Phosphate dysregulation via the XPR1-KIDINS220
protein complex is a therapeutic vulnerability in ovarian cancer. Nat Cancer. 2022 Jun;3(6):681-695.
https://doi.org/10.1038/s43018-022-00360-7 |
| 27 | Wang Y, Yang W, Pu Q, Yang Y, Ye S, Ma Q. The effects and mechanisms of SLC34A2 in tumorigenesis
and progression of human non-small cell lung cancer. J Biomed Sci 2015;22(1):52.
https://doi.org/10.1186/s12929-015-0158-7 |
| 28 | Li Y, Chen X, Lu H. Knockdown of SLC34A2 Inhibits Hepatocellular Carcinoma Cell Proliferation and
Invasion. Oncol Res. 2016;24(6):511-519.
https://doi.org/10.3727/096504016X14719078133483 |
| 29 | Bao Z, Chen L, Guo S. Knockdown of SLC34A2 inhibits cell proliferation, metastasis, and elevates
chemosensitivity in glioma. J Cell Biochem 2019;120(6):10205-10214.
https://doi.org/10.1002/jcb.28305 |
| 30 | Rangel LBA, Sherman-Baust CA, Werbyj RP, Schwartz DR, Cho KR, Morin PJ. Characterization of novel
human ovarian cancer-specific transcripts (HOSTs) identified by serial analysis of gene expression.
Oncogene 2003;22:7225-7232.
https://doi.org/10.1038/sj.onc.1207008 |
| 31 | Soares IC, Simões K, de Souza JE, Okamoto OK, Wakamatsu A, Tuma M. In silico analysis and
immunohistochemical characterization of NaPi2b protein expression in ovarian carcinoma with
monoclonal antibody Mx35. Appl Immunohistochem Mol Morphol 2012;20(2):165-172.
https://doi.org/10.1097/PAI.0b013e318228e232 |
| 32 | Akasu-Nagayoshi Y, Hayashi T, Kawabata A, Shimizu N, Yamada A, Yokota N. PHOSPHATE exporter
XPR1/SLC53A1 is required for the tumorigenicity of epithelial ovarian cancer. Cancer Sci
2022;113(6):2034-2043.
https://doi.org/10.1111/cas.15358 |
| 33 | Russo-Abrahão T, Lacerda-Abreu MA, Gomes T, Cosentino-Gomes D, Carvalho-de-Araújo AD, Rodrigues
MF. Characterization of inorganic phosphate transport in the triple-negative breast cancer cell
line, MDA-MB-231. PLoS One 2018;13(2):e0191270.
https://doi.org/10.1371/journal.pone.0191270 |
| 34 | Yang K, Li Z, Chen Y, Yin F, Ji X, Zhou J. Na, K-ATPase α1 cooperates with its endogenous ligand
to reprogram immune microenvironment of lung carcinoma and promotes immune escape. Sci Adv
2023;9(6):eade5393.
https://doi.org/10.1126/sciadv.ade5393 |
| 35 | Shiozaki A, Miyazaki H, Niisato N, Nakahari T, Iwasaki Y, Itoi H. Furosemide, a blocker of
Na+/K+/2Cl cotransporter, diminishes proliferation of poorly differentiated human gastric cancer
cells by affecting G0/G1 state. J Physiol Sci 2006;56(6):401-406.
https://doi.org/10.2170/physiolsci.RP010806 |
| 36 | Chang SH, Yu KN, Lee YS, An GH, Beck GR Jr, Colburn NH. Elevated inorganic phosphate stimulates
Akt-ERK1/2-Mnk1 signaling in human lung cells. Am J Respir Cell Mol Biol 2006;35(5):528-539.
https://doi.org/10.1165/rcmb.2005-0477OC |
| 37 | Xiao Y, Meng C, Lin J, Huang C, Zhang X, Long Y. Ouabain targets the Na+/K+-ATPase α3 isoform to
inhibit cancer cell proliferation and induce apoptosis. Oncol Lett 2017;14(6):6678-6684.
https://doi.org/10.3892/ol.2017.7070 |
| 38 | Fujii T, Shimizu T, Yamamoto S, Funayama K, Fujita K, Tabuchi Y. Crosstalk between Na+,K+-ATPase
and a volume-regulated anion channel in membrane microdomains of human cancer cells. Biochim Biophys
Acta Mol Basis Dis 2018;1864(11):3792-3804.
https://doi.org/10.1016/j.bbadis.2018.09.014 |
| 39 | Michaels AM, Zoccarato A, Hoare Z, Firth G, Chung YJ, Kuchel PW. Disrupting Na+ ion homeostasis
and Na+/K+ ATPase activity in breast cancer cells directly modulates glycolysis in vitro and in
vivo. Cancer Metab 2024;12(1):15.
https://doi.org/10.1186/s40170-024-00343-5 |
| 40 | Hu J, Li G, Liu Z, Ma H, Yuan W, Lu Z. Bicarbonate transporter SLC4A7 promotes EMT and metastasis
of HNSCC by activating the PI3K/AKT/mTOR signaling pathway. Mol Carcinog 2023;62(5):628-640.
https://doi.org/10.1002/mc.23511 |
| 41 | Li CL, Yang R, Sun Y, Feng Y, Song YB. N58A Exerts Analgesic Effect on Trigeminal Neuralgia by
Regulating the MAPK Pathway and Tetrodotoxin-Resistant Sodium Channel. Toxins (Basel)
2021;13(5):357.
https://doi.org/10.3390/toxins13050357 |
| 42 | Jin H, Xu CX, Lim HT, Park SJ, Shin JY, Chung YS. High dietary inorganic phosphate increases lung
tumorigenesis and alters Akt signaling. Am J Respir Crit Care Med 2009;179(1):59-68.
https://doi.org/10.1164/rccm.200802-306OC |