Original Article - DOI:10.33594/000000819
Accepted 11 September 2025 - Published
online 14 October 2025
Allopurinol Attenuates Senescence and Oxidative Stress in Endothelial Cells Exposed to Serum from Hypertensive Patients with Hyperuricemia - a Pilot Study
Keywords
Abstract
Background/Aims:
Endothelial cell senescence is a key contributor to the development of vascular pathologies, including arterial hypertension. Uric acid has been shown to promote oxidative stress and inflammation, thereby accelerating endothelial dysfunction and senescence. Although xanthine oxidase inhibition with allopurinol has demonstrated cardiovascular benefits, its effect on endothelial senescence remains insufficiently characterised. This study aimed to investigate the impact of sera from patients with arterial hypertension and elevated uric acid levels on the senescence of human umbilical vein endothelial cells, and to determine whether allopurinol treatment modulates this effect.Materials:
In this study, human umbilical vein endothelial cells were cultured and exposed to sera from hypertensive patients with elevated uric acid levels (≥5 mg/dL) before and after six weeks of allopurinol treatment (300 mg/day). A control group consisting of healthy individuals with normal uric acid levels was established. Eighteen participants of both sexes were recruited to the study. Markers of senescence (SA-β-Gal, γ-H2A.X, 53BP1), oxidative stress (mitochondrial and cytosolic reactive oxygen species, mitochondrial mass, membrane potential), cell proliferation and inflammatory cytokine production were analysed.Results:
Sera from hypertensive patients before treatment induced endothelial senescence and oxidative stress significantly and altered secretory profiles in endothelial cells compared to the controls; however, allopurinol treatment led to a partial reversal of these changes. Specifically, it reduced mitochondrial reactive oxygen species, mitochondrial mass and γ-H2A.X levels, and increased cell proliferation. However, not all markers returned to baseline values, and some (e.g. inflammatory mediators) remained elevated or even increased further after treatment.Conclusion:
The findings demonstrate that allopurinol partially reverses uric acid- and hypertension-related endothelial senescence and oxidative damage. However, the incomplete normalisation suggests that multiple overlapping pathways contribute to vascular cell senescence in this context.Introduction
Recent findings have highlighted that the senescence of vascular cells, particularly endothelial cells, is a critical factor contributing to the development of several vascular pathologies, including atherosclerosis, arterial hypertension (AH), and broader cardiovascular diseases (CVDs) [1, 2].
Uric acid (UA), especially in its increased concentrations, induces endothelial dysfunction and is a cardiovascular (CV) risk factor, including for AH [3]. Endothelial injury can be triggered by reactive oxygen species (ROS), generated during UA synthesis under the hypoxic conditions associated with CVDs, due to the elevated activity of the enzyme xanthine oxidase (XO)[4, 5]. Hence, despite being an antioxidant, UA cannot neutralise all the ROS generated during its production with the participation of XO [6]. Additionally, high concentrations of UA itself contribute to endothelial damage [3]. Increased cellular stress and the damage it causes lead to cellular senescence [7].
In the AH patient group, elevated UA values are more prevalent than in the general population [8, 9]. The results of the RISK study indicate that in Poland, the incidence of hyperuricemia in the cohort of untreated AH patients is approximately 15% [10]. In the older population, this number rises to 23% in individuals over 65, and to as much as 30% in individuals over 90 [11]. Worldwide data show that more than 75, 5 million people suffer from hyperuricemia [12]. Thus, the observed relationship holds significant meaning from an epidemiological perspective. Consequently, there is a necessity for a thorough investigation into this subject and the role of UA in the pathophysiology of AH, as well as the search for those drugs capable of inhibiting or reversing the harmful effects of UA.
Allopurinol is an XO inhibitor that lowers UA synthesis. To date, numerous studies have shown a correlation between allopurinol usage and cardiovascular (CV) risk, and overall mortality risk reduction [13-15]. Reduction of UA concentration by allopurinol in patients with high CV risk was found to be beneficial [16].
The decrease in CVD risk resulted from the ability of allopurinol to improve endothelial function, particularly its action mechanism related to the inhibition of XO [5, 6,13-15, 17]. A study by MacIsaac et al. demonstrated that allopurinol therapy, particularly at higher doses (≥300 mg daily), was associated with a reduced incidence of stroke and cardiovascular events in older individuals with AH (high CV risk)[16].
The Pressioni Arteriose Monitorate e Loro Associazioni (PAMELA) study was one of the first to consider UA as a CV risk factor. In a group of 2045 patients observed for 16 years, an increase in UA by 1 mg/dL significantly increased the risk of developing AH. A 1 mg/dL increase in serum UA concentration was associated with a 22% increase in the risk of CV death and a 12% increase in the risk of death from all causes. The serum UA concentration best predicting death from CV causes was 5.4 mg/dL (sensitivity 61%, specificity 67%), and 4.9 mg/dL for death from all causes (sensitivity 68%, specificity 55%)[18].
In vitro studies on the role of allopurinol in endothelial cell senescence are somewhat scarce. Added to human umbilical vein endothelial cells (HUVECs) subjected to hypoxia, this compound inhibited its angiogenic potential [19]. However, Polytarchou et al. did not observe a significant effect of allopurinol on HUVECs’ proliferation, migration, or nitric oxide (NO) synthase activity [20]. Furthermore, there is a paucity of studies explaining allopurinol's impact on vascular endothelial cell senescence [21-23].
In our study, we aimed to evaluate how the sera of patients with AH and elevated UA levels affect the senescence process of HUVECs. Furthermore, to gain a deeper understanding of the role of allopurinol treatment in our patients, we aimed to assess how allopurinol treatment (via certain sera) influences HUVECs’ senescence.
Materials and Methods
Materials
Unless otherwise stated, all reagents were purchased from Sigma-Aldrich Corp. (St. Louis, MO).
Patients
Recruitment of participants, clinical assessment and allopurinol treatment.
Examination of the patients and the laboratory test needed to classify patients to control or intervention
group
were performed during hospitalisation in the Department of Hypertensiology, Angiology and Internal Disease
at
Poznan University of Medical Sciences.
Patients were informed about the benefits and risks of intervening with allopurinol. The study was not
blinded.
The criterion for the drug’s inclusion was the UA concentration level in the serum, and a placebo was not
planned.
Before starting the study, each patient was familiarised with the written information about the study
(including
information about the study, procedures, risks, data use and the right to withdraw from the study). Only
people
who gave informed written consent were included in the study.
The bioethics commission approved the study with Poznań University of Medical Sciences, resolution No.
954/19, 3
October, 2019.
Eighteen participants of both sexes were recruited to the study. The study group consisted of thirteen
patients
with a serum UA concentration ≥ 5 mg/dL and well-controlled AH, who received allopurinol at a dose of 300
mg per
day. Blood sera were taken from all participants before (V1) each treatment and after (V2) six weeks of
pharmacotherapy. The dose and the time were determined based on previous research [13, 24-26] The UA
concentration was selected according to expert recommendations based on the results of the PAMELA study
[18]. A
serum concentration of UA above 5 mg/dL determined the decision to perform allopurinol intervention. The
control
groups consisted of five healthy volunteers without AH and with a serum UA concentration below 5mg/dL.
HUVECs were cultured for in vitro tests. The cells were exposed to sera of study group V1, study
group V2
and the control group.
Blood samples (36 ml) were taken from a forearm vein in a standard ambulatory procedure.
Patient recruitment
Patients willing to participate and fulfilling the inclusion and exclusion criteria were enrolled in the
study.
Inclusion criteria: age: 18–65 years, with well-controlled hypertension according to the 2018 European
Society
of Cardiology/European Society of Hypertension (ESC/ESH) guidelines (office blood pressure lower than
140/90
mmHg) [8].
Exclusion criteria: diagnosis of secondary hypertension; chronic diseases including diabetes, other CVDs
(e.g.
coronary heart disease, heart failure, atherosclerosis); autoimmune diseases; acute inflammation process;
renal
impairment (glomerular filtration rate lower than 60 ml/min/1, 72m2); thyroid disease (e.g.
hyperthyroidism,
hypothyroidism, thyroiditis cancer; storage disease, cancer history; gout; Body Mass Index (BMI) over 30
kg/m2;
taking medications that affect UA concentration (including allopurinol and diuretics, except other
hypotensive
drugs); women taking oestrogen preparations, pregnant or lactating.
Cell culture and exposure to patients’ sera
HUVECs were obtained from Lonza (Walkersville, MD, USA). They were cultured using EBM™-2 Basal Medium
supplemented with EGM™-2 SingleQuots™ (Lonza). The cell cultures were maintained at 37°C in a humidified
environment containing 95% air and 5% CO₂.
During the experiments, HUVECs were seeded in culture dishes at a high density (80–90% confluency) and
simultaneously exposed to 80% of patients’ sera, collected both before and after treatment. Exposure to
patients’ sera lasted for 168 hours.
The exposure protocol was based on exposing HUVECs to various concentrations of patient serum.
Subsequently, a
preliminary MTT assay was performed, from which a serum concentration of 80% was selected for further
experiments.
Conditioned medium (CM) generation
To obtain a CM, young HUVECs were plated in 25 cm² flasks. Once they reached around 80% confluency, they
were
exposed to patients’ sera for 168 hours. Following this exposure, the cells were washed with PBS and then
incubated in a serum-free medium for 72 hours. After this period, the media were collected, and the cells
responsible for producing them were counted. All collected CM were filtered using a 0.2 μm pore size
filter and
stored at -80°C until further use.
Detection of senescence-associated β-Galactosidase (SA-β-Gal)
The expression of SA-β-Gal in cell cultures was visualised using the method described earlier [27]. The
activity
of SA-β-Gal in cell extracts was determined by assessing the conversion rate of
4-methylumbelliferyl-β-D-galactopyranose to 4-methylumbelliferone, following the procedure outlined by
Gary and
Kindell [28]. The quantification of activity was performed using a Synergy™ 2 spectrofluorometer (BioTek
Instruments, Winooski, VT, USA).
Detection of histone γ-H2A.X and 53BP1
Immunofluorescence of γ-H2A.X and 53BP1 foci was assessed using the methods outlined in [29]with
anti-γ-H2A.X
(Ser139) and anti-53BP1 antibodies (Novus Biologicals, Abingdon, UK). Images of the immunoreactions were
captured using the Axio Vert.A1 microscope (Carl-Zeiss, Jena, Germany), and the fluorescence intensity of
γ-H2A.X and 53BP1 foci was quantified using the Synergy™ H1 spectrofluorometer.
Cell proliferation measurements
Cell proliferation was assessed by counting cells using a Bürker chamber. Cells were plated at a
predetermined
low density and cultured for 168 hours, and then harvested and counted.
Cell secretome and the levels of selected parameters directly in patients’ sera
The concentrations of IL-6, IL-8, Vascular Endothelial Growth Factor (VEGF), angiopoietin,
Platelet-Derived
Growth Factor-DD (PDGF-DD), and Transforming Growth Factor Beta 1 (TGF-β1) in CM from HUVECs and the
concentrations of IL-6, CXCL8/IL-8, VEGF and TGF-β1 in patients’ sera were measured using the appropriate
DuoSet® Immunoassay Development kits (R&D Systems), following the manufacturer's protocol.
Measurement of oxidative stress-related parameters
To evaluate mitochondrial superoxide and cellular peroxide generation, mitochondrial ROS (MitoSOX Red -
Mitochondria-targeted Hydroethidine-based Superoxide Indicator) and dihydrorhodamine 123 (DHR) were
utilised.
Mitochondrial mass was assessedbystaining with 10-n-nonyl-acridine orange (NAO). Furthermore, the
mitochondrial
membrane potential (ΔΨm) was measured in cells stained with 1 μM 5, 5′,6, 6′-tetrachloro-1, 1′,3,
3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1). Detailed methodologies for these analyses were
described
in [30].
Statistical Analysis
The statistical analysis was conducted using GraphPad Prism 10.00 software (GraphPad Software, San Diego,
USA).
A Student’s t-test was used to compare two groups. Where necessary, Welch’s t-test or the Mann–Whitney
test was
applied. For paired data, a paired Student’s t-test or the Wilcoxon signed-rank test was used when
appropriate.
Results are presented as mean ± standard deviation (SD), and differences with a P-value of less than 0.05
were
considered statistically significant.
Results
Two experimental groups were compared: HUVECs exposed to serum from hypertensive patients with UA (sUA) levels equal to or greater than 5 mg/dL (study group–V1), and HUVECs exposed to serum from the same patients after 6 weeks of treatment with allopurinol (study group–V2). The control group consisted of HUVECs exposed to serum from healthy patients with sUA levels below 5 mg/dL. The characteristics of all groups of patients are presented in Table 1.
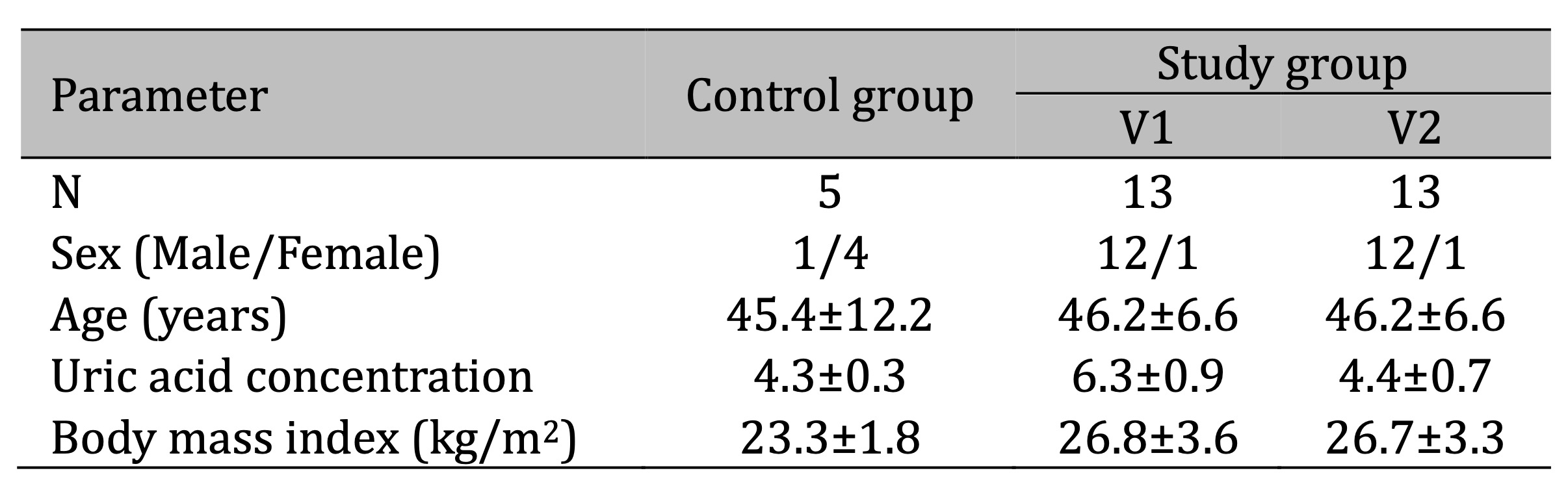
Table 1:
Serum from study group V1, compared to the control group, worsened all the tested markers in HUVECs significantly. An increase of SA-β-Gal, γ-H2A.X and 53BP1, and a decrease in proliferation were observed. Serum from patients before allopurinol treatment worsened all oxidative stress-related parameters in HUVECs significantly. A notable increase in cytosolic and mitochondrial reactive oxygen species (ROS) and changes associated with mitochondrial damage were observed. The results are presented in Figures 1 and 2.
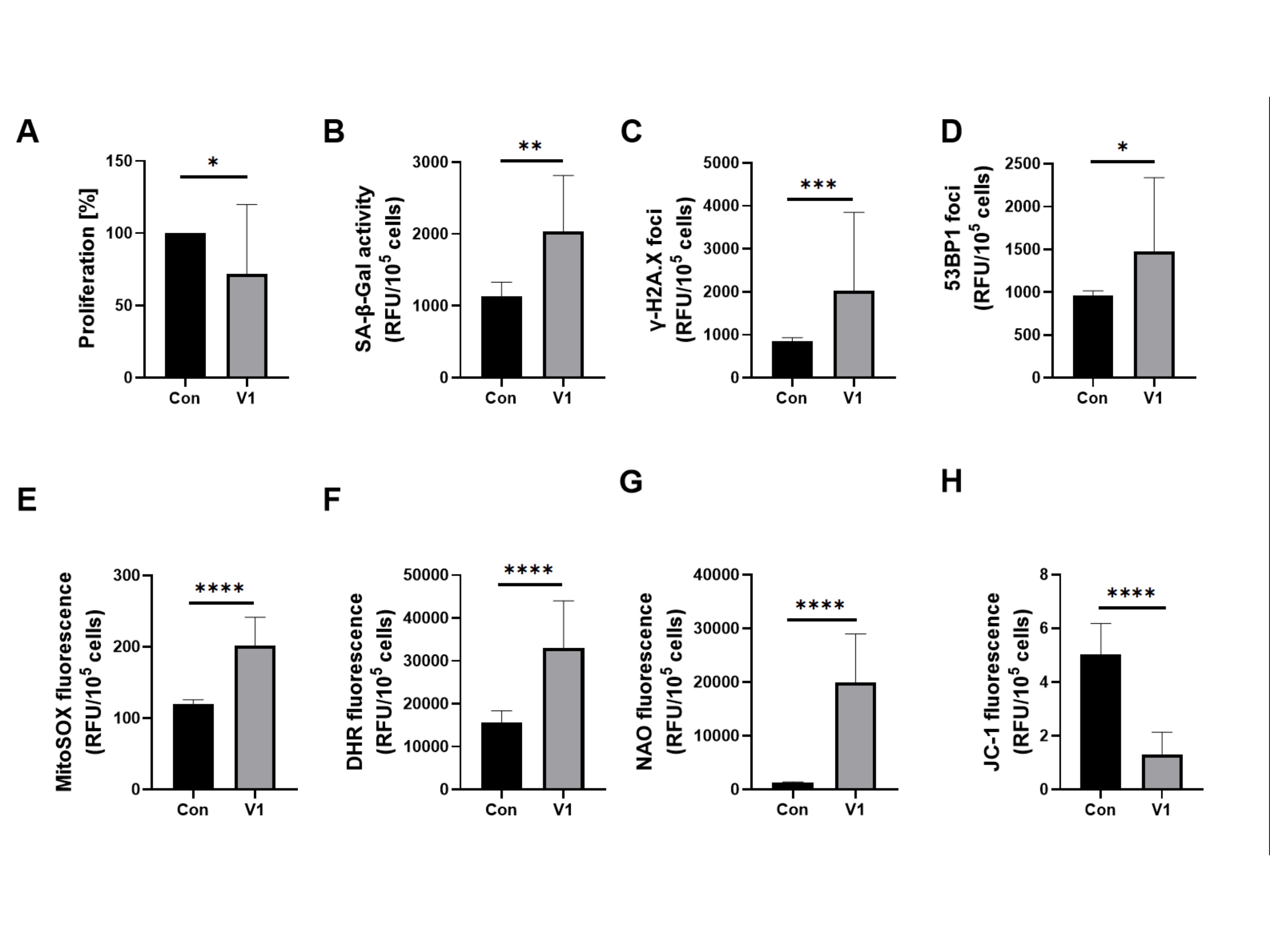
Fig. 1: Effect of patient serum from study group V1 versus that from the control group on (a) proliferation, senescence markers induction and oxidative stress-related parameters in HUVECs. Quantification (b) of SA-β-Gal activity, (c) expression of γ-H2A.X, (d) expression of 53BP1 of HUVECs, (e) mitochondrial superoxides, (f) cellular peroxides, (g) mitochondrial mass, and (h) mitochondrial membrane potential in HUVECs. Results are presented as mean ± SD. Statistical significance: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****).
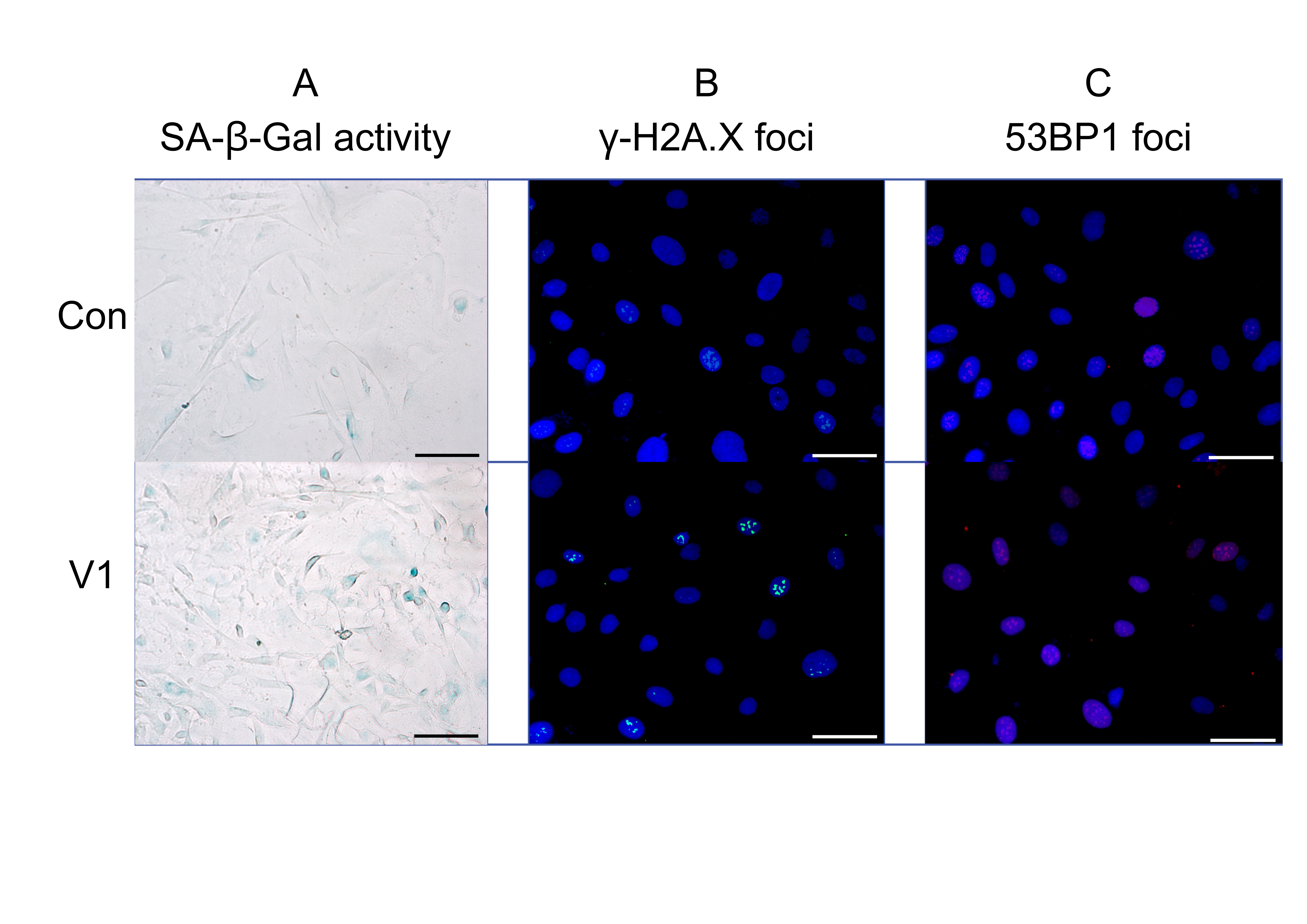
Fig. 2: Effect of patient serum from study group V1 compared to the control group: representative fluorescence images showing (a) SA-β-Gal activity, (b) γ-H2A.X expression, and (c) 53BP1 expression. The uploaded image includes a scale bar and appropriate magnification: scale bar = 500 µm, magnification = 40×.
Serum from patients’ study group – V2 treatment increased HUVECs proliferation significantly. Among the senescence markers, it reduced γ-H2A.X significantly while SA-β-Gal and 53BP1 levels remained unchanged. A significant decrease in MitoSOX red and mitochondrial mass (NAO) was also observed, but cellular peroxides (DHR) and mitochondrial membrane potential values (JC-1) were as before treatment. The results are presented in Figures 3 and 4.
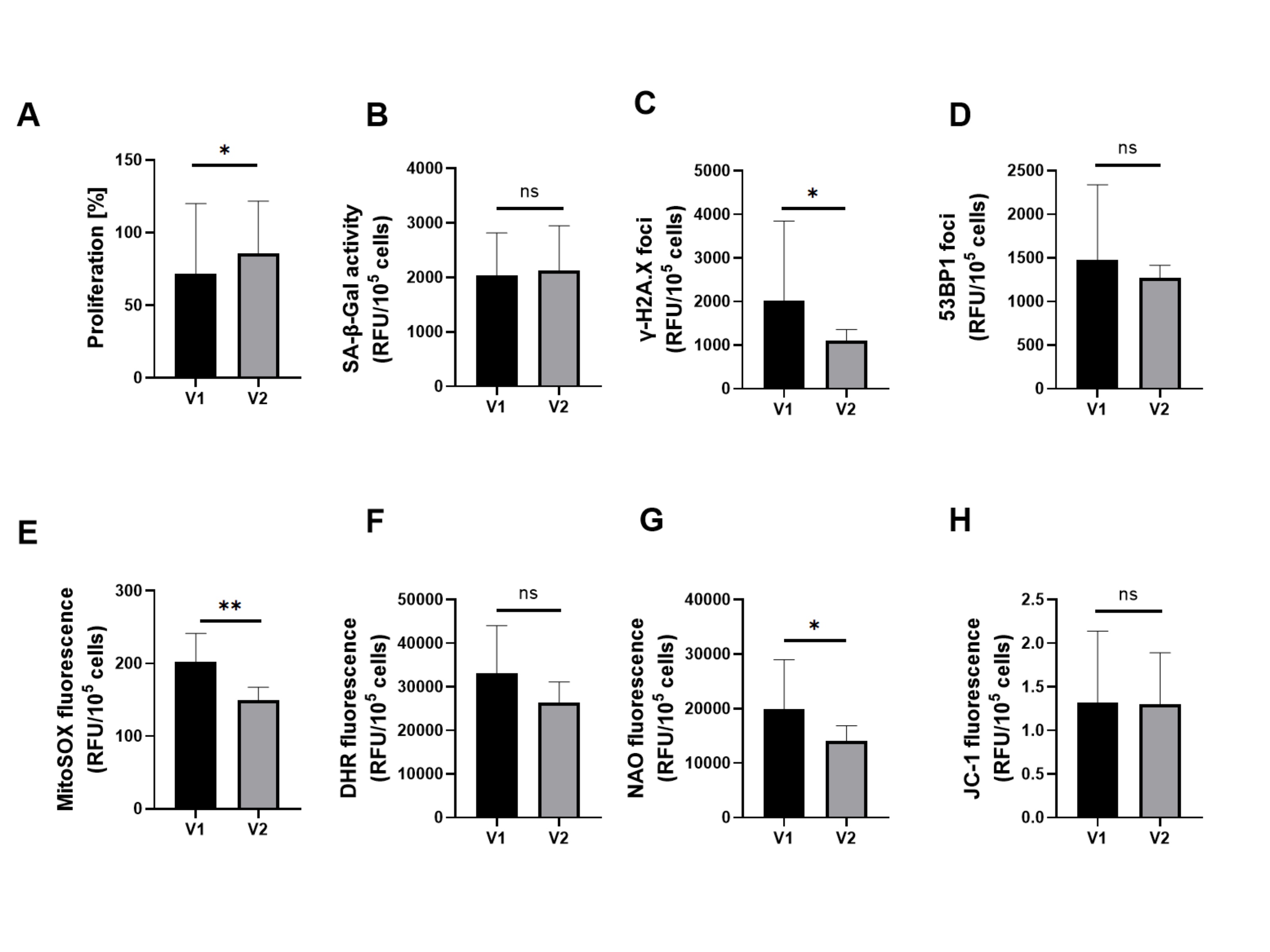
Fig. 3: Effect of patient serum from study group V1 versus study group V2 on (a) proliferation, senescence markers induction and oxidative stress-related parameters in HUVECs. Quantification of (b) SA-β-Gal activity, (c) expression of γ-H2A.X, (d) expression of 53BP1, of HUVECs, (e) mitochondrial superoxides, (f) cellular peroxides, (g) mitochondrial mass, and (h) mitochondrial membrane potential in HUVECs. Results are presented as mean ± SD. Statistical significance: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****).
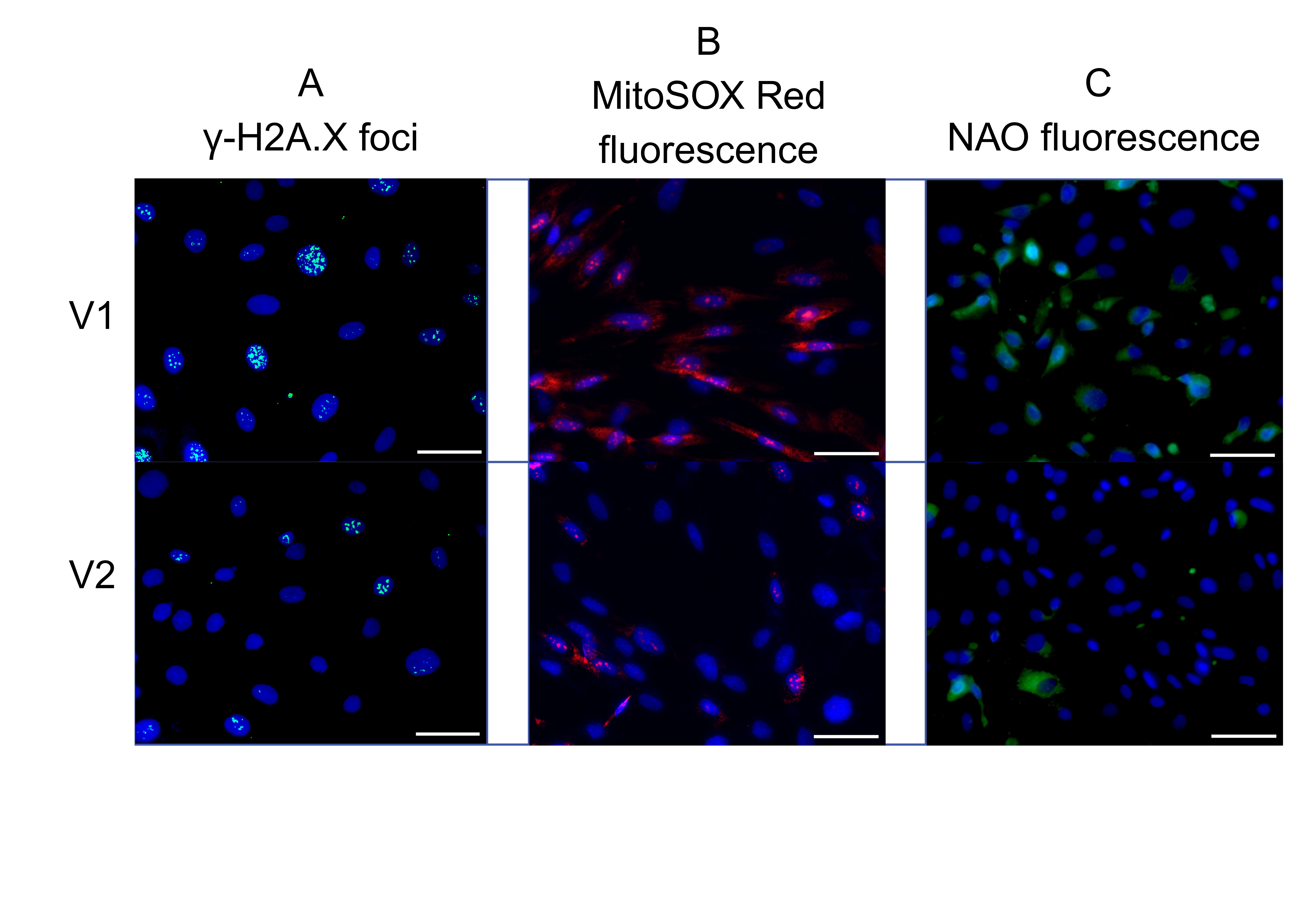
Fig. 4: Effect of patient serum from study group V1 versus study group V2: representative fluorescence images showing (a) γ-H2A.X expression, (b) mitochondrial superoxide levels, and (c) mitochondrial mass. The uploaded image includes a scale bar and appropriate magnification: scale bar = 500 µm, magnification = 40×.
Four parameters representing SASP were selected arbitrarily, and their levels were measured in the patient’s serum. Compared to the control group, a significant increase was observed in all parameters: IL-6, IL-8, TGF-β, and VEGF in the ‘study group V1’ group. After six weeks of allopurinol therapy, a significant increase was noted for IL-6, a significant decrease for VEGF, and a non-significant reduction for IL-8 and TGF-β. The results are presented in Fig. 5.
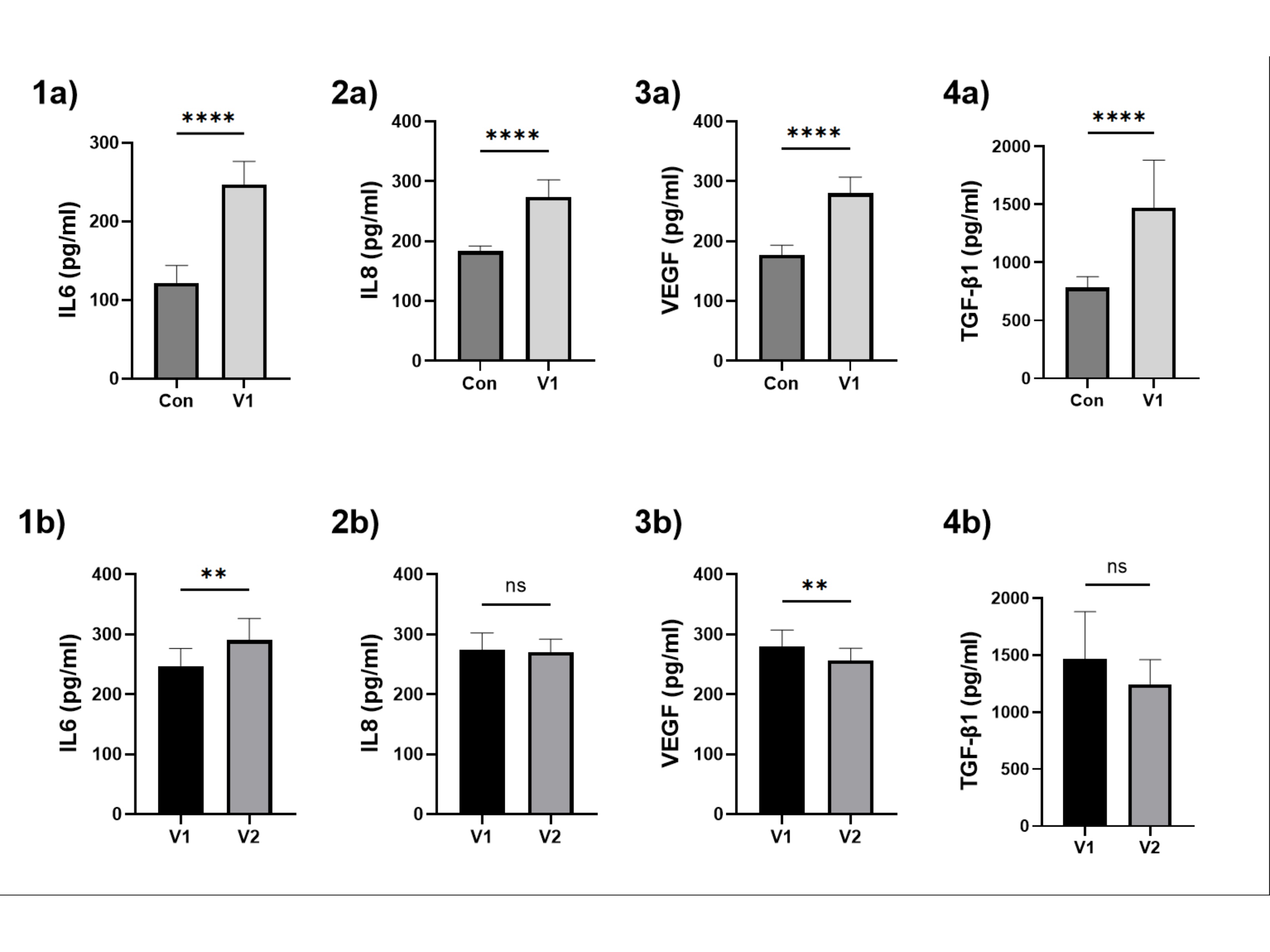
Fig. 5: The assessment of selected inflammatory cytokines and growth factors in patients’ sera. (1) IL-6, (2) IL-8, (3) VEGF, (4) TGF-β1. Results are presented as mean ± SD. a – for the comparison between the control group and the study group V1; b – for the comparison between study group V1 and study group V2. Statistical significance: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****).
After exposing HUVECs to the patients’ serum, a CM was collected, in which IL-6, IL-8, TGF-β1, VEGF, angiopoietin and PDGF-DD were assessed. A significant parameter increase was observed when comparing HUVECs exposed to the control serum with those in the study group V1.
When comparing HUVECs exposed to the serum from patients V1 with those from V2, a significant decrease in VEGF, angiopoietin and PDGF-DD was noted, alongside a significant increase in TGF-β1, IL-6 and non-significant in IL-8. The results are presented in Fig. 6.
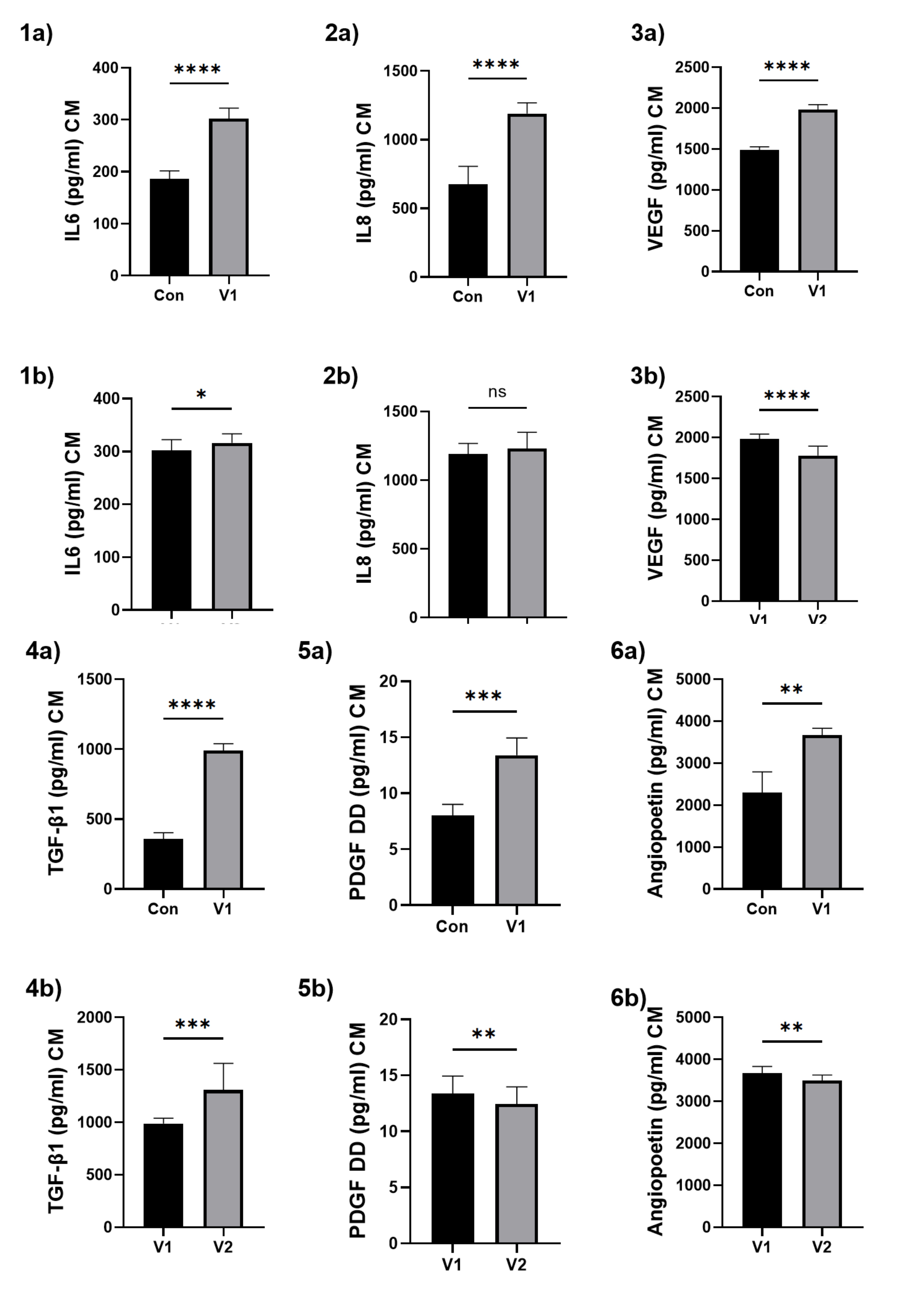
Fig. 6: The assessment of selected inflammatory cytokines and growth factors produced by HUVECs after patients’ serum exposition. (1) IL-6, (2) IL-8, (3) VEGF, (4) TGF-β1, (5) PDGF DD, (6) angiopoietin. a – for the comparison between the control group and the study group V1; b – for the comparison between study group V1 and study group V2. Results are presented as mean ± SD. Statistical significance: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****).
Discussion
Our objective was to examine the effect of allopurinol treatment in hypertensive patients with a serum UA concentration ≥ 5 mg/dL (via specific serum exposure) on HUVEC senescence. The analysis of the results obtained reveals clear trends. The serum from patients before allopurinol treatment exhibited pro-senescence properties on HUVECs. The serum, compared to the control group, had a significantly negative impact on all senescence-assessed parameters (γ-H2A.X, SA-β-gal, 53BP1) and proliferation in HUVECs. The tested serum (V1) also significantly increased all oxidative stress parameters (MitoSOX red, DHR, NAO, JC-1). The results obtained are consistent with previously published studies. Researchers believe that both UA and AH can induce cellular senescence. They explain this phenomenon by the increased production of ROS associated with both factors, which negatively impacts endothelial cells and promotes cellular senescence [21-23, 31,32].
The serum from the V2 study group (after allopurinol therapy) partially reversed some of the observed changes in HUVECs, but not all of them. It can be suggested that allopurinol, to some extent, exerts an antioxidant and anti-senescence effect on HUVECs. It improved proliferation significantly and reduced γ-H2A.X, which is a marker of senescence-associated DNA damage and the activation of the so-called DNA damage response [33]. A similar effect of allopurinol was recently reported by Wang et al., who used a chemically induced rat model of asthma, where XO inhibition by allopurinol attenuated oxidative stress and DNA damage [34]. Likewise, Klein et al. showed that allopurinol influenced DNA synthesis and repair processes, indicating its potential to support cellular recovery following damage [35].
Allopurinol treatment in our study reduced mitochondrial ROS production significantly and decreased mitochondrial mass in HUVECs. In the case of serum V1, we observed an increase in mitochondrial mass, reflecting increased mitochondrial biogenesis. One may speculate that it represents a compensatory cell response to mitochondrial damage or a functional impairment and is often accompanied by elevated ROS production, as a higher number of mitochondria means more active respiratory chains, which are the main source of ROS [36, 37]. Therefore, allopurinol, by reducing mitochondrial biogenesis, decreases mitochondrial-derived free radicals, undoubtedly exerting a beneficial and protective effect on HUVECs. Similar observations have been reported by other researchers, demonstrating that allopurinol exerts antioxidant effects by reducing ROS production in rat cardiomyocytes and in the plasma of horses [38, 39]. Rus et al. showed in a rat model that XO activity can occur in mitochondria and is inhibited by allopurinol. This implies that the generation of mitochondrial ROS is not due exclusively to the damage of the respiratory chain but also involves XO’s catalytic function [40]. Other authors have already highlighted the positive impact of allopurinol on mitochondrial metabolism. Gladden et al. demonstrated that cardiomyocytes subjected to 3 hours of cyclic stretching exhibited increased XO activity and mitochondrial swelling, which was prevented by pre-treatment with allopurinol [41]. Similarly to our findings, both NAO and mitochondrial ROS levels decreased after allopurinol treatment. Nakano et al. also showed that allopurinol, in dogs’ liver tissue exposed to warm ischemia, helped prevent the decline in mitochondrial ATP metabolism and reduced the production of lipid peroxides, which contributed to the quick recovery of mitochondrial redox balance [42].
However, in our study, even after allopurinol treatment, some markers of senescence and oxidative stress were not fully reversed. The partial reversal of cellular senescence and oxidative stress markers, particularly the reduction of γ-H2A.X, mitochondrial ROS (MitoSOX red), and NAO without a significant impact on 53BP1, SA-β-Gal, cytosolic ROS (DHR) or mitochondrial membrane potential (JC-1) suggests that inhibition of XO alone is insufficient to fully reverse the damaging effects induced by UA and/or AH. This may indicate that other sources of oxidative stress and inflammatory pathways not directly related to XO could contribute to our patients’ cellular damage. Both AH and elevated UA levels contribute to increased intracellular ROS production. The underlying mechanisms are complex and multifactorial, which is likely the primary reason for the incomplete improvement of the evaluated parameters following allopurinol treatment.
The increase in ROS within cells is an element inseparable from AH. AH promotes the development of inflammation and ROS production. Elevated vascular pressure exerted on the walls of blood vessels induces mechanical stress, activating endothelial and smooth muscle cells to produce increased levels of ROS [43]. Activation of the renin-angiotensin-aldosterone system (RAAS) contributes significantly to organ damage and ROS generation in AH[44]. Chronically elevated blood pressure contributes to vascular remodelling [45]. This is likely driven by angiotensin II-induced activation of growth factors such as a platelet-derived growth factor (PDGF), a vascular endothelial growth factor (VEGF), and a fibroblast growth factor (FGF) [45]. The pathophysiology of AH is consistent with our findings. Evaluating selected SASP-related parameters in patients’ serum and in the CM produced by HUVECs after serum exposure aligns with the above. When comparing Con and V1, VEGF and PDGF DD—measured both in patient serum and CM—would be expected to decrease alongside reduced proliferation; however, our results reveal a paradoxical increase in thes parameters. The observed increase is most likely due to strong RAAS activation induced by elevated UA levels in AH. The decrease in VEGF levels in serum and VEGF and PDGF in a conditioned medium in the ‘after allopurinol’ group supports the researchers’ theory that UA may activate the RAAS. The reduction after XO inhibition probably results from allopurinol lowering UA, which reduces UA-dependent RAAS activation [21, 45, 46].
RAAS activation promotes an increase in cellular ROS. Angiotensin II stimulates ROS production via AT1-mediated NADPH oxidase activation and enhances inflammation through AP-1 and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways [47-51]. NADPH oxidase can also be activated directly by UA. Moreover, UA promotes inflammation by suppressing AMPK and activating pathways such as Rho kinase, NF-κB, and Janus Kinase 2/Signal Transducer and Activator of Transcription 3 (JAK2/STAT3) [52].
A key source of ROS in patients with elevated UA and AH is ROS generated during UA production itself. Hypoxia and inflammation activate XO, leading to UA synthesis accompanied by ROS generation in the final steps [53]. In hypoxia, mitochondrial lysis releases enzymes that irreversibly convert dehydrogenase to XO [54]. Although UA can inhibit XO retroactively, ROS production still accompanies these reactions [55]. As described above, XO is just the tip of the iceberg—many other factors contribute to ROS generation and senescence, making the mechanism more complex than it seems, but allopurinol cannot inhibit all sources of ROS production.
Further analysis of our results suggests the involvement of additional mechanisms of allopurinol action or lack thereof. Allopurinol has been shown to inhibit Hypoxia-Inducible Factor 1- α (HIF1-α), as observed by other researchers, which is also evident in our study [56). Typically, angiopoietin levels rise due to HIF1-α activation in response to hypoxia. Hypoxia and inflammation accompanying AH and UA in group V1 promote increased angiopoietin levels. Then, allopurinol inhibits HIF-1, reducing angiopoietin production, as observed in patients’ serum (group V2) and CM from HUVECs treated with this serum.
Some researchers suggest that allopurinol may inhibit the NF-κB pathway, thereby reducing IL-6 levels and inflammation [57]. In our results, IL-6 was elevated considerably in the serum of patients in the study groups (before and after allopurinol treatment) and in the CM of HUVECs exposed to these sera. Furthermore, in the serum of patients after treatment, IL-6 levels increased significantly in both cases. An explanation for this may be provided by the study of Pearlstein et al. They observed a significant rise in IL-6 after subjecting HUVECs to hypoxia. The addition of a NAD(P)H oxidase inhibitor (apocynin), an XO inhibitor (allopurinol), or a nitric oxide synthase inhibitor (N-nitro-l-arginine) did not reduce IL-6 levels significantly [58]. Hypoxia accompanying AH is likely too strong a factor to reveal the inhibitory effect of allopurinol on the NF-κB pathway/IL-6.
Allopurinol does not affect IL-8 or TGF-β1. The level of IL-8 increased significantly both in the serum and in the CM. Similar associations have been observed by other researchers in patients with AH and elevated UA levels [59, 60]. Allopurinol did not affect IL-8 levels significantly. Multiple factors may contribute to its activation, not solely XO. Researchers suggest that it plays a key role in driving senescence in patients with CVDs [61]. TGF-β1 levels are elevated in patients with AH, as confirmed in our study: in group V1, TGF-β1 increased both in serum and CM[62]. Allopurinol treatment reduced its serum levels, but the change was not statistically significant. It is possible that, in our patients, the TGF-β1-driven senescence pathway is too strong to be reversed by XO inhibition and the resulting ROS reduction. Moreover, HUVECs exposed to patient sera secreted significantly more TGF-β1 into CM, even after treatment, suggesting that the serum retains its pro-senescent properties despite allopurinol therapy.
The lack of reduction, and in some cases even an increase, of IL-6, IL-8, or TGF-β1 in serum and/or CM after allopurinol treatment underscores the complexity and multifactorial nature of the underlying mechanisms. In patients with AH and hyperuricemia, levels of IL-6, IL-8, and TGF-β1 are elevated, contributing to the chronic inflammation associated with these conditions. These cytokines activate common signalling pathways, including NF-κB, MAPK (Mitogen-Activated Protein Kinase), or JAK/STAT, and engage in cross-talk, mutually enhancing their expression. TGF-β1 additionally promotes a profibrotic and pro-inflammatory endothelial phenotype, while ROS generated within this cascade further amplifies cytokine production, leading to increased oxidative stress and tissue fibrosis [63-64]. Blocking a single pathway is often insufficient, as inflammatory signals are sustained through multiple, interconnected mechanisms. Allopurinol inhibits XO, reducing UA and ROS production; however, it cannot suppress cytokines activated through XO-independent pathways.
One could speculate whether UA or AH is the more significant factor influencing the results obtained. On one hand, UA may contribute to the development of AH, while on the other, it could be a consequence of it. To confirm this, large, well-designed studies are needed. International experts on the diagnosis and treatment of patients with hyperuricaemia and high CV risk recommended considering allopurinol for individuals with UA levels above 5 mg/dL who have at least two of the following risk factors: AH, diabetes, dyslipidaemia, previous stroke, myocardial infarction, or chronic kidney disease. They also suggested targeting a UA level below 5 mg/dL [12]. However, recently, results from the large ALL-HEART study were published, showing that allopurinol did not provide benefits in secondary prevention for patients with ischemic heart disease [65-66]. Our findings may offer a partial explanation as to why the ALL-HEART study did not yield the outcomes anticipated by the authors. It is possible that the patients in the ALL-HEART study were too ill to benefit from allopurinol treatment. The more severely ill the patients, with extensive vascular damage, the more factors that could contribute to the induction of cellular senescence. In our study, patients had lower CV risk than those in the ALL-HEART trial and did not present with significant organ damage. The results of the ongoing multicentre, randomised, double-blind ALL-VASCOR trial will be of particular importance. This study aims to assess the benefits of allopurinol therapy in patients with high and very high CV risk. Patients with ischemic heart disease have been excluded, and the study protocol excluded most of the limitations previously raised regarding the ALL-HEART trial [67].
Conclusion
Our study highlights the role of oxidative stress and mitochondrial dysfunction in endothelial senescence. The clinical implications of our study suggest that allopurinol may be beneficial for patients with AH and elevated UA levels. However, appropriate antihypertensive therapy must also be ensured. Additionally, our analysis indicates that treatments targeting the inflammatory pathways described above could also benefit this patient group.
The present study represents a pilot translational investigation, and the findings may provide guidance for other researchers and serve as a basis for generating hypotheses for larger controlled trials.
Limitations
Undoubtedly, a key limitation of this study is the small sample size and sex imbalance. However, finding such a refined group of patients with only AH and elevated UA levels, without other comorbidities, is challenging. During the pre-screening phase in our department, patients underwent a comprehensive diagnostic evaluation, which enabled the precise selection of the study group. Another aspect is the UA levels in the patients. In the ‘before allopurinol’ group, the average serum UA level was 6.3 mg/dL. Our results might have been more spectacular if the patients had started treatment with serum UA levels above 7–7.5 mg/dL. However, elevated UA levels are often associated with the onset of additional conditions, and in our case, such patients generally had a BMI exceeding 30 kg/m², which is an excluded criterion. Additionally, the study used 80% serum, which may have further diluted the UA levels to which the HUVECs were exposed.
Another consideration is the allopurinol dose—300 mg/day. A higher dose might have yielded better results, but for our patient group (these are patients who have moderate CVD risk), obtaining ethics committee approval and ensuring patient compliance could have been more challenging. Moreover, the duration of allopurinol administration (6 weeks) may have been too short to reverse all the adverse effects of the long-term impact of elevated UA and AH on the body. Lastly, working with serum samples presents limitations, as the parameters we assessed represent only a small fraction of the complex interactions within the serum.
As a single-center, short-term, non-randomized, and non-blinded study without a placebo control, the potential for clinical extrapolation is limited.
Acknowledgements
Author Contributions
All authors contributed to the study conception and design. Material preparation, data collection and
analysis
were performed by Katarzyna Lewandowska, Paweł Uruski, Krzysztof Książek and Justyna Mikua-Pietrasik. The
first
draft of the manuscript was written by Katarzyna Lewandowska and all authors commented on previous
versions of
the manuscript. All authors read and approved the final manuscript.
Funding Sources
This research was funded by statutory funds of the Department of Hypertensiology, Angiology and Internal
Medicine, Poznan University of Medical Sciences, Poland.
Statement of Ethics
The bioethics commission approved the study with Poznań University of Medical Sciences, resolution No.
954/19, 3
October, 2019. Before starting the study, each patient was familiarised with the written information about
the
study (including information about the study, procedures, risks, data use and the right to withdraw from
the
study). Only people who gave informed written consent were included in the study.
Disclosure Statement
The authors have no conflicts of interest to declare.
References
| 1 | Bai B, Liang Y, Xu C, Lee MYK, Xu A, Wu D, Vanhoutte PM, Wang Y: Cyclin-dependent kinase
5-mediated hyperphosphorylation of sirtuin-1 contributes to the development of endothelial
senescence and atherosclerosis.Circulation. 2012 Aug 7;126(6):729-40.
https://doi.org/10.1161/CIRCULATIONAHA.112.118778 |
| 2 | Buford TW: Hypertension and aging. Ageing Res Rev. 2016;26:96-111.
https://doi.org/10.1016/j.arr.2016.01.007 |
| 3 | Maruhashi T, Hisatome I, Kihara Y, Higashi Y: Hyperuricemia and endothelial function: From
molecular background to clinical perspectives. Atherosclerosis.Elsevier Ireland Ltd;
2018;278:226-31.
https://doi.org/10.1016/j.atherosclerosis.2018.10.007 |
| 4 | Farquharson CAJ, Butler R, Hill A, Belch JJF, Struthers AD: Allopurinol improves endothelial
dysfunction in chronic heart failure. Circulation. 2002 Jul 9;106(2):221-6.
https://doi.org/10.1161/01.CIR.0000022140.61460.1D |
| 5 | Prasad M, Matteson EL, Herrmann J, Gulati R, Rihal CS, Lerman LO, Lerman A: Uric Acid Is
Associated with Inflammation, Coronary Microvascular Dysfunction, and Adverse Outcomes in
Postmenopausal Women. Hypertension. 2017 Feb 1;69(2):236-42.
https://doi.org/10.1161/HYPERTENSIONAHA.116.08436 |
| 6 | Becker BF: Towards the physiological function of uric acid. Free Radic Biol Med.
1993;14(6):615-31.
https://doi.org/10.1016/0891-5849(93)90143-I |
| 7 | Lewandowska K, Mikuła-Pietrasik J, Książek K, Tykarski A, Uruski P: Uric Acid Promotes Human
Umbilical Vein Endothelial Cell Senescence In vitro. Metabolites. 2025 Jun 14;15(6):402.
https://doi.org/10.3390/metabo15060402 |
| 8 | Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, Clement DL, Coca A, Simone
G, Dominiczak A, Kahan T, Mahfoud F, Redon J, Ruilope L, Zanchetti A, Kerins M, Kjeldsen SE, Kreutz
R, Laurent S, Lip GYH, McManus R, Narkiewicz K, Ruschitzka F, Schmieder RE, Shlyakhto E, Tsioufis C,
Aboyans V, Desormaiset I: 2018 ESC/ESH Guidelines for the management of arterial hypertensionThe
Task Force for the management of arterial hypertension of the European Society of Cardiology (ESC)
and the European Society of Hypertension (ESH). Eur Heart J. 2018 Sep 1;39(33):3021-104.
https://doi.org/10.1201/9780429199189-75 |
| 9 | Cannon PJ, Stason WB, Demartini FE, Sommers SC, Laragh JH: Hyperuricemia in Primary and Renal
Hypertension. N Engl J Med.1966 Sep 16;275(9):457-64.
https://doi.org/10.1056/NEJM196609012750902 |
| 10 | Kostka-Jeziorny K, Tykarski A: Relationship between hyperuricemia and other cardiovascular risk
factors in patients with essential, untreated arterial hypertension in the population of RISK study.
Nadcisnienie Tetnicze. 2008;12(3):190-9.
|
| 11 | Winder M, Owczarek AJ, Mossakowska M, Broczek K, Grodzicki T, Wierucki Ł, Chudek J: Prevalence of
Hyperuricemia and the Use of Allopurinol in Older Poles-Results from a Population-Based PolSenior
Study. Int J Environ Res Public Health. 2021 Jan 2;18(2):1-14.
https://doi.org/10.3390/ijerph18020387 |
| 12 | Borghi C, Domienik-Karłowicz J, Tykarski A, Widecka K, Filipiak KJ, Jaguszewski MJ, Narkiewicz K,
Mancia G: Expert consensus for the diagnosis and treatment of patient with hyperuricemia and high
cardiovascular risk: 2021 update. Cardiol J. 2021;28(1):1-14.
https://doi.org/10.5603/CJ.a2021.0001 |
| 13 | Wei L, Fahey T, Struthers AD, MacDonald TM: Association between allopurinol and mortality in heart
failure patients: A long-term follow-up study. Int J Clin Pract. 2009;63(9):1327-33.
https://doi.org/10.1111/j.1742-1241.2009.02118.x |
| 14 | Dubreuil M, Zhu Y, Zhang Y, Seeger JD, Lu N, Rho YH, Choi HK: Allopurinol initiation and all-cause
mortality in the general population. Ann Rheum Dis. 2015 Jul 1;74(7):1368-72.
https://doi.org/10.1136/annrheumdis-2014-205269 |
| 15 | Søltoft Larsen K, Pottegård A, Lindegaard HM, Hallas J: Effect of Allopurinol on Cardiovascular
Outcomes in Hyperuricemic Patients: A Cohort Study. Am J Med. 2016;129:299-306.
https://doi.org/10.1016/j.amjmed.2015.11.003 |
| 16 | MacIsaac RL, Salatzki J, Higgins P, Walters MR, Padmanabhan S, Dominiczak AF, Touyz RM, Dawson J:
Allopurinol and Cardiovascular Outcomes in Adults with Hypertension. Hypertension. 2016 Mar
1;67(3):535-40.
https://doi.org/10.1161/HYPERTENSIONAHA.115.06344 |
| 17 | Maiuolo J, Oppedisano F, Gratteri S, Muscoli C, Mollace V: Regulation of uric acid metabolism and
excretion. Int J Cardiol. 2016;213:8-14.
https://doi.org/10.1016/j.ijcard.2015.08.109 |
| 18 | Bombelli M, Ronchi I, Volpe M, Facchetti R, Carugo S, Dell'Oro R, Cuspidi C, Grassi G, Mancia G:
Prognostic value of serum uric acid: New-onset in and out-of-office hypertension and long-term
mortality. J Hypertens. 2014;32(6):1237-44.
https://doi.org/10.1097/HJH.0000000000000161 |
| 19 | Sun Y, George J, Rocha S: Dose-Dependent Effects of Allopurinol on Human Foreskin Fibroblast Cells
and Human Umbilical Vein Endothelial Cells under Hypoxia. PLoS One. 2015 Apr 1;10(4):e0123649.
https://doi.org/10.1371/journal.pone.0123649 |
| 20 | Polytarchou C, Papadimitriou E: Antioxidants inhibit human endothelial cell functions through
down-regulation of endothelial nitric oxide synthase activity. Eur J Pharmacol. 2005;510(1-2):31-8.
https://doi.org/10.1016/j.ejphar.2005.01.004 |
| 21 | Yu MA, Sánchez-Lozada LG, Johnson RJ, Kang DH: Oxidative stress with an activation of the
renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric
acid-induced endothelial dysfunction. J Hypertens. 2010;28(6):1234-42.
https://doi.org/10.1097/HJH.0b013e328337da1d |
| 22 | Li Y, Zhao L, Qi W: Uric acid, as a double-edged sword, affects the activity of epidermal growth
factor (EGF) on human umbilical vein endothelial cells by regulating aging process. Bioengineered.
2022;13(2):3877-95.
https://doi.org/10.1080/21655979.2022.2027172 |
| 23 | Wu G, Liu J, Ma G, Wei Q, Song X: Hyperuricemia Facilitates Uric Acid-Mediated Vascular
Endothelial Cell Damage by Inhibiting Mitophagy. Cell Biochem Biophys. 2024;83:811-821.
https://doi.org/10.1007/s12013-024-01512-5 |
| 24 | Feig DI, Kang DH, Johnson RJ. Medical progress: Uric acid and cardiovascular risk. Vol. 359, New
England Journal of Medicine. Massachussetts Medical Society; 2008. p. 1811-21.
https://doi.org/10.1056/NEJMra0800885 |
| 25 | Noman A, Ang DS, Ogston S, Lang CC, Struthers AD. Effect of high-dose allopurinol on exercise in
patients with chronic stable angina: a randomised, placebo controlled crossover trial. Lancet.
2010;375(9732):2161-7.
https://doi.org/10.1016/S0140-6736(10)60391-1 |
| 26 | Muir SW, Harrow C, Dawson J, Lees KR, Weir CJ, Sattar N, et al. Allopurinol Use Yields Potentially
Beneficial Effects on Inflammatory Indices in Those With Recent Ischemic Stroke A Randomized,
Double-Blind, Placebo-Controlled Trial. 2008.
https://doi.org/10.1161/STROKEAHA.108.519793 |
| 27 | Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I,
Pereira-Smith O: A biomarker that identifies senescent human cells in culture and in aging skin in
vivo. Proc Natl Acad Sci U S A. 1995 Sep 26;92(20):9363.
https://doi.org/10.1073/pnas.92.20.9363 |
| 28 | Gary RK, Kindell SM: Quantitative assay of senescence-associated beta-galactosidase activity in
mammalian cell extracts. Anal Biochem. 2005 Aug 15;343(2):329-34.
https://doi.org/10.1016/j.ab.2005.06.003 |
| 29 | Mikuła-Pietrasik J, Sosińska P, Murias M, Michalak M, Wierzchowski M, Piechota M, Sikora E,
Książek K: Resveratrol Derivative, 3, 3',4, 4'-Tetrahydroxy-trans-Stilbene, Retards Senescence of
Mesothelial Cells via Hormetic-Like Prooxidative Mechanism. J Gerontol A Biol Sci Med Sci. 2015 Nov
19;70(10):1169-80.
https://doi.org/10.1093/gerona/glu172 |
| 30 | Mikuła-Pietrasik J, Uruski P, Pakuła M, Maksin K, Szubert S, Woźniak A, Naumowicz E, Szpurek D,
Tykarski A, Książek K: Oxidative stress contributes to hepatocyte growth factor-dependent
pro-senescence activity of ovarian cancer cells. Free Radic Biol Med. 2017 Sep 1;110:270-9.
https://doi.org/10.1016/j.freeradbiomed.2017.06.015 |
| 31 | Harman D: Free radical theory of aging. Mutat Res. 1992 Sep 1;275(3-6):257-66.
https://doi.org/10.1016/0921-8734(92)90030-S |
| 32 | Afsar B, Afsar RE: Hypertension and cellular senescence. Biogerontology. 2023 Aug 1;24(4):457-78.
https://doi.org/10.1007/s10522-023-10031-4 |
| 33 | Carracedo J, Ramírez-Carracedo R, Alique M, Ramírez-Chamond R: Endothelial Cell Senescence in the
Pathogenesis of Endothelial Dysfunction. Endothel Dysfunct - Old Concepts New Challenges.
InTech;2018;p.10.
https://doi.org/10.5772/intechopen.73024 |
| 34 | Wang Y, Le Y, Wu J, Zhao W, Zhang Q, Xu G, Gong Z, Xu, MaY1, Yu Ch, Cai S, Zhao H: Inhibition of
xanthine oxidase by allopurinol suppresses HMGB1 secretion and ameliorates experimental asthma.
Redox Biol. 2024 Apr 1;70:103021.
https://doi.org/10.1016/j.redox.2023.103021 |
| 35 | Klein G, Wottawa A, Rainer F: The effect of uricosuric and uricostatic drugs on DNA metabolism.
Acta Med Austriaca. 1976;3(3):91-7.
|
| 36 | Correia‐Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J,
Merz A, Rushton MD, CharlesM, Jurk D, Tait SWG, Czapiewski R, Greaves L, Nelson G, Bohlooly-Y M,
Rodriguez-Cuenca S, Vidal-Puig A, Mann D, Saretzki G, Quarato G, Green DR, Adams PD, Zglinicki T,
Korolchuk VI, Passos JF: Mitochondria are required for pro‐ageing features of the senescent
phenotype. EMBO J. 2016 Apr 4;35(7):724-42.
https://doi.org/10.15252/embj.201592862 |
| 37 | Finkel T, Holbrook NJ: Oxidants, oxidative stress and the biology of ageing. Nat 2000 4086809 2000
Nov 9;408(6809):239-47.
https://doi.org/10.1038/35041687 |
| 38 | Kang SM, Lim S, Song H, Chang W, Lee S, Bae SM, Chung JH, Lee H, Kim HG, Yoon DH, Kim TW, Jang Y,
Sung JM, Chung NS, Hwanget KCh: Allopurinol modulates reactive oxygen species generation and Ca 2+
overload in ischemia-reperfused heart and hypoxia-reoxygenated cardiomyocytes. Eur J Pharmacol. 2006
Mar 27;535(1-3):212-9.
https://doi.org/10.1016/j.ejphar.2006.01.013 |
| 39 | Mills PC, Smith NC, Harris RC, Harris P: Effect of allopurinol on the formation of reactive oxygen
species during intense exercise in the horse. Res Vet Sci. 1997;62(1):11-6.
https://doi.org/10.1016/S0034-5288(97)90172-7 |
| 40 | Rus DA, Sastre J, Viña J, Pallardó F V: Induction of mitochondrial xanthine oxidase activity
during apoptosis in the rat mammary gland. Front Biosci. 2007;12(4):1184-9.
https://doi.org/10.2741/2136 |
| 41 | Gladden JD, Zelickson BR, Wei CC, Ulasova E, Zheng J, Ahmed MI, Chen Y, Bamman M, Ballinger S,
Darley-Usmar V, Dell'Italia LJ: Novel insights into interactions between mitochondria and xanthine
oxidase in acute cardiac volume overload. Free Radic Biol Med. 2011 Dec 1;51(11):1975-84.
https://doi.org/10.1016/j.freeradbiomed.2011.08.022 |
| 42 | Nakano M, Sugano M, Terasaki M, Morimoto T, Mashima S, Mitsuyoshi A, Sasaki H , Kumada K, Ozawa K:
Preserved mitochondrial function by allopurinol despite deteriorated hemodynamics in warm
ischemia-damaged canine liver. Res Exp Med (Berl). 1992 Dec;192(6):389-99.
https://doi.org/10.1007/BF02576296 |
| 43 | Rodrigo R, González J, Paoletto F: The role of oxidative stress in the pathophysiology of
hypertension. Hypertens Res. 2011 Jan 13;34(4):431-40.
https://doi.org/10.1038/hr.2010.264 |
| 44 | Duprez DA: Role of the renin-angiotensin-aldosterone system in vascular remodeling and
inflammation: a clinical review. J Hypertens. 2006;24(6):983-91.
https://doi.org/10.1097/01.hjh.0000226182.60321.69 |
| 45 | Cohuet G, Struijker-Boudier H. Mechanisms of target organ damage caused by hypertension:
therapeutic potential. Pharmacol Ther. 2006 Jul;111(1):81-98.
https://doi.org/10.1016/j.pharmthera.2005.09.002 |
| 46 | Perlstein TS, Gumieniak O, Hopkins PN, Murphey LJ, Brown NJ, Williams GH, Hollenberg NK, Fisheret
NDL: Uric acid and the state of the intrarenal renin-angiotensin system in humans. Kidney Int.
2004;66(4):1465-70.
https://doi.org/10.1111/j.1523-1755.2004.00909.x |
| 47 | Rajagopalan S, Kurz S, Münzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DH: Angiotensin
II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH
oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996 Apr
15;97(8):1916-23.
https://doi.org/10.1172/JCI118623 |
| 48 | Ma J, Li Y, Yang X, Liu K, Zhang X, Zuo X, Ye R, Wang Z, Shi R, Meng Q, Chen X: Signaling pathways
in vascular function and hypertension: molecular mechanisms and therapeutic interventions. Signal
Transduct Target Ther 2023 81. 2023 Apr 20;8(1):1-30.
https://doi.org/10.1038/s41392-023-01430-7 |
| 49 | Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, Namba M: Inhibitory effects of
antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and
angiotensin II. Circulation. 1998 Aug 15;98(8):794-9.
https://doi.org/10.1161/01.CIR.98.8.794 |
| 50 | Rodríguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ: Oxidative stress, renal infiltration
of immune cells, and salt-sensitive hypertension: All for one and one for all. Am J Physiol - Ren
Physiol. 2004;286(4):F606-16.
https://doi.org/10.1152/ajprenal.00269.2003 |
| 51 | Das UN, Sciences UNDL Is angiotensin-II an endogenous pro-inflammatory molecule ?
2005;11(5):155-62.
|
| 52 | Spiga R, Marini MA, Mancuso E, Di Fatta C, Fuoco A, Perticone F, Andreozzi F, Mannino GCh, Sesti
G: Uric Acid Is Associated with Inflammatory Biomarkers and Induces Inflammation Via Activating the
NF-κB Signaling Pathway in HepG2 Cells. Arterioscler Thromb Vasc Biol. 2017 Jun 1;37(6):1241-9.
https://doi.org/10.1161/ATVBAHA.117.309128 |
| 53 | Hink HU, Santanam N, Dikalov S, McCann L, Nguyen AD, Parthasarathy S, Harrison DG, Fukaiet T:
Peroxidase properties of extracellular superoxide dismutase: role of uric acid in modulating in vivo
activity. Arterioscler Thromb Vasc Biol. 2002 Sep 1;22(9):1402-8.
https://doi.org/10.1161/01.ATV.0000027524.86752.02 |
| 54 | Saksela M, Lapatto R, Raivio KO: Irreversible conversion of xanthine dehydrogenase into xanthine
oxidase by a mitochondrial protease. FEBS Lett. 1999 Jan 29;443(2):117-20.
https://doi.org/10.1016/S0014-5793(98)01686-X |
| 55 | Nishino T: The conversion of xanthine dehydrogenase to xanthine oxidase and the role of the enzyme
in reperfusion injury. Vol. 116, Journal of Biochemistry. Oxford University Press; 1994 p.1-6.
https://doi.org/10.1093/oxfordjournals.jbchem.a124480 |
| 56 | Abe H, Semba H, Takeda N: The Roles of Hypoxia Signaling in the Pathogenesis of Cardiovascular
Diseases. J Atheroscler Thromb. 2017;24(9):884.
https://doi.org/10.5551/jat.RV17009 |
| 57 | Schlesinger N, Brunetti L: Beyond urate lowering: Analgesic and anti-inflammatory properties of
allopurinol. Semin Arthritis Rheum. 2020 Jun 1;50(3):444-50.
https://doi.org/10.1016/j.semarthrit.2019.11.009 |
| 58 | Pearlstein DP, Ali MH, Mungai PT, Hynes KL, Gewertz BL, Schumacker PT: Role of mitochondrial
oxidant generation in endothelial cell responses to hypoxia. Arterioscler Thromb Vasc Biol. 2002 Apr
1;22(4):566-73.
https://doi.org/10.1161/01.ATV.0000012262.76205.6A |
| 59 | Tanase DM, Gosav EM, Radu S, Ouatu A, Rezus C, Ciocoiu M, Costea CF, Floria M: Arterial
Hypertension and Interleukins: Potential Therapeutic Target or Future Diagnostic Marker? Int J
Hypertens. 2019 May 2;2019:3159283.
https://doi.org/10.1155/2019/3159283 |
| 60 | Estevez-Garcia IO, Gallegos-Nava S, Vera-Pérez E, Silveira LH, Ventura-Ríos L, Vancini G,
Hernández-Díaz C, Sánchez-Muñoz F, Ballinas-Verdugo MA, Gutierrez M, Pineda C, Rodriguez-Henriquez
P, Castillo-Martínez D, Amezcua-Guerra LM: Levels of Cytokines and MicroRNAs in Individuals With
Asymptomatic Hyperuricemia and Ultrasonographic Findings of Gout: A Bench-to-Bedside Approach.
Arthritis Care Res. 2018 Dec 1;70(12):1814-21.
https://doi.org/10.1002/acr.23549 |
| 61 | Tominaga K, Suzuki HI: TGF-β Signaling in Cellular Senescence and Aging-Related Pathology. Int J
Mol Sci. 2019 Oct 2;20(20):5002.
https://doi.org/10.3390/ijms20205002 |
| 62 | Suthanthiran M, Li B, Song JO, Ding R, Sharma VK, Schwartz JE, August P: Transforming growth
factor-beta 1 hyperexpression in African-American hypertensives: A novel mediator of hypertension
and/or target organ damage. Proc Natl Acad Sci U S A. 2000 Mar 28;97(7):3479-84.
https://doi.org/10.1073/pnas.97.7.3479 |
| 63 | Xu Z, Yang S, Tan Y, Wang H, Tao J, Liu Q, Wang Q, Feng W, Li Z, Wang Ch, Cui L: Inflammation in
cardiovascular-kidney-metabolic syndrome: key roles and underlying mechanisms-a comprehensive
review. Mol Cell Biochem (2025). https://doi.org/10.1007/s11010-025-05379-9
https://doi.org/10.1007/s11010-025-05379-9 |
| 64 | Ageeva T, Rizvanov A, Mukhamedshina Y: NF-κB and JAK/STAT Signaling Pathways as Crucial Regulators
of Neuroinflammation and Astrocyte Modulation in Spinal Cord Injury. Cells. 2024 Mar 26;13(7):581.
https://doi.org/10.3390/cells13070581 |
| 65 | MacKenzie IS, Ford I, Walker A, Hawkey C, Begg A, Avery A, Taggar J, Wei L, Struthers AD,
MacDonald TM: Multicentre, prospective, randomised, open-label, blinded end point trial of the
efficacy of allopurinol therapy in improving cardiovascular outcomes in patients with ischaemic
heart disease: Protocol of the ALL-HEART study. BMJ Open. 2016;6(9):1-8.
https://doi.org/10.1136/bmjopen-2016-013774 |
| 66 | Mackenzie IS, Hawkey CJ, Ford I, Greenlaw N, Pigazzani F, Rogers A, Struthers AD, Begg AG, Wei L,
Avery AJ, Taggar JS, Walker A, Duce SL, Barr RJ, Dumbleton JS, Rooke ED, Townend JN, Ritchie LD,
MacDonald TM: Allopurinol versus usual care in UK patients with ischaemic heart disease (ALL-HEART):
a multicentre, prospective, randomised, open-label, blinded-endpoint trial. Lancet.
2022;400(10359):1195-205.
|
| 67 | Lewandowska K, Lipski D, Uruski P, Narkiewicz K, Januszewicz A, Wolf J, Prejbisz A, Rajzer M,
Więcek A, Tykarskiet A. Randomised, double-blind, placebo-controlled study evaluating the effect of
allopurinol on the risk of cardiovascular events in patients with high and very high cardiovascular
risk, including the presence of long-COVID-19 syndrome: the ALL-VASCOR study protocol. BMJ Open.
2024;14(7):1-8.
https://doi.org/10.1136/bmjopen-2023-075741 |